O Brasil é um dos dez maiores mercados de dispositivos médicos do mundo
Este artigo faz parte do Guia Definitivo de Engenharia Biomédica.
Marco Regulatório Reformulado: As Principais RDCs
O arcabouço regulatório brasileiro para dispositivos médicos foi completamente renovado a partir de 2022. A pedra angular dessa reforma é a RDC 751/2022
| RDC | Ano | Tema |
|---|---|---|
| RDC 751 | 2022 | Norma-pilar: classificação e registro de dispositivos médicos |
| RDC 665 | 2022 | Boas Práticas de Fabricação (BPF), alinhamento à ISO 13485 |
| RDC 657 | 2022 | SaMD, Software como Dispositivo Médico |
| RDC 830 | 2023 | Dispositivos para diagnóstico in vitro (IVDs) |
| RDC 837 | 2023 | Investigações clínicas com dispositivos médicos |
| RDC 848 | 2024 | Requisitos Essenciais de Segurança e Desempenho (RESD) |
| RDC 850 | 2024 | CBPF via MDSAP: validade ampliada de 2 para 4 anos |
| RDC 982 | 2025 | Inteligência artificial na certificação de BPF |
Esse conjunto normativo posiciona o Brasil como uma das agências regulatórias mais estruturadas para dispositivos médicos entre países em desenvolvimento, com crescente harmonização com FDA, Health Canada, TGA e MHLW. Para entender em profundidade a organização interna da agência e suas atribuições, consulte nosso artigo sobre como funciona a ANVISA: estrutura, atribuições e seu papel na regulamentação.
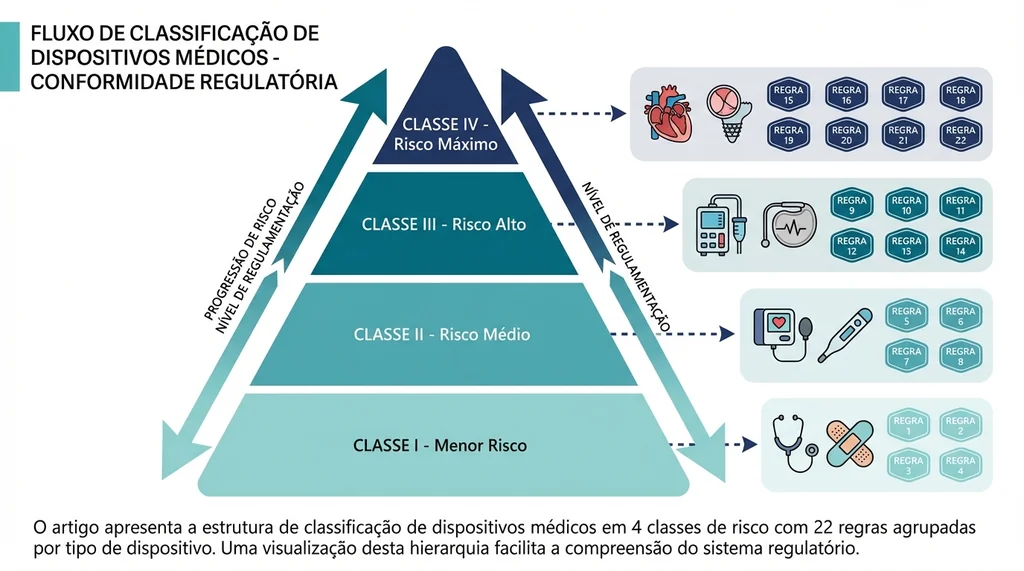
As 4 Classes de Risco e as 22 Regras de Classificação
A RDC 751/2022 organiza os dispositivos médicos em quatro classes de risco, determinadas pela aplicação de 22 regras de classificação distribuídas em quatro grupos temáticos.
Grupos de regras de classificação
- Regras 1 a 4: dispositivos não invasivos (contato com pele, mucosas, feridas)
- Regras 5 a 8: dispositivos invasivos (cirúrgicos, de longa duração, implantáveis)
- Regras 9 a 13: dispositivos ativos (elétricos, emissão de radiação, diagnóstico ativo)
- Regras 14 a 22: regras especiais (contracepção, desinfecção, nanomateriais, SaMD, IA)
Três regras especiais merecem atenção específica para o contexto atual:
Descrição completa da imagem
Diagrama das 4 classes de risco de dispositivos médicos ANVISA com exemplos por categoria regulatória brasileira.
- Regra 11 (SaMD): classifica softwares como dispositivos médicos em Classes I a IV conforme a gravidade do dano potencial ao paciente
- Regra 19 (Nanomateriais): determina classificação em Classe III ou IV para dispositivos que utilizam nanotecnologia
- Regra 22: aplica-se a dispositivos terapêuticos ativos com diagnóstico integrado, como sistemas combinados de monitoramento e terapia
Tabela resumo das classes
| Classe | Nível de Risco | Via Regulatória | Validade | CBPF Obrigatório | Exemplos |
|---|---|---|---|---|---|
| I | Baixo | Notificação | Indefinida | Não | Instrumentos cirúrgicos simples, luvas de procedimento |
| II | Médio | Notificação | Indefinida | Não | Bombas de infusão, cadeiras de rodas, curativos avançados |
| III | Alto | Registro | 10 anos | Sim | Dialisadores, ventiladores pulmonares, implantes ortopédicos |
| IV | Máximo | Registro | 10 anos | Sim | Stents coronarianos, marca-passos, próteses articulares |
A classificação correta é o ponto de partida de toda a estratégia regulatória. Um erro de classificação, pode resultar em embargo do produto, multas e necessidade de refazer todo o processo de registro.
Fluxo de Registro: Do Protocolo à Concessão
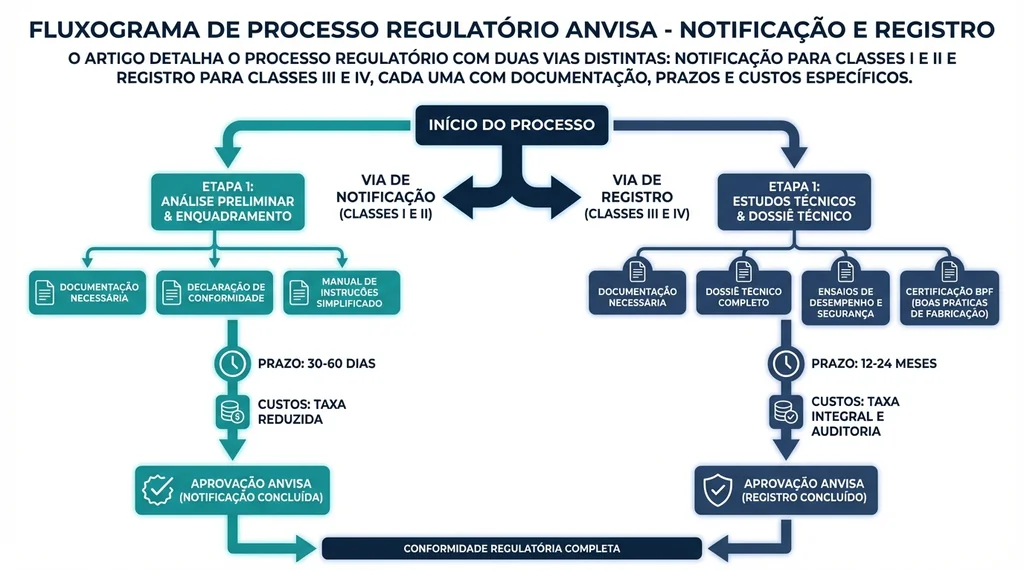
O processo regulatório para dispositivos médicos no Brasil se divide em duas vias principais: notificação (Classes I e II) e registro (Classes III e IV). Cada via tem requisitos, prazos e custos distintos.
Via de Notificação (Classes I e II)
A notificação é um processo simplificado, adequado para produtos de menor risco. O fabricante ou importador cadastra o produto no sistema DATAVISA da ANVISA, comprova conformidade com os requisitos técnicos e obtém a notificação, que tem validade por prazo indefinido enquanto o produto mantiver conformidade regulatória.
Descrição completa da imagem
Fluxograma do processo de registro de dispositivos médicos na ANVISA com etapas de notificação e registro por classe de risco.
O prazo estimado para conclusão é de aproximadamente 30 dias corridosR$ 1.406 em taxas ANVISA (Taxa de Fiscalização de Vigilância Sanitária, TFVS).
Via de Registro (Classes III e IV)
O registro é o processo mais rigoroso, exigido para produtos de alto e máximo risco. Inclui análise técnica detalhada pela ANVISA, exigência de Certificado de Boas Práticas de Fabricação (CBPF) e, para determinadas categorias, dados clínicos do produto.
Os documentos centralmente exigidos incluem:
- Dossiê técnico completo (sumário técnico, especificações, rotulagem)
- Relatório de avaliação de desempenho clínico ou equivalência técnica
- Relatório de gestão de riscos (ISO 14971)
- Certificado de Boas Práticas de Fabricação (CBPF) válido
- Autorização de Funcionamento de Empresa (AFE) para fabricantes nacionais
- Laudos de ensaios de segurança e desempenho conforme normas aplicáveis
Os prazos e custos para registro variam conforme a classe:
| Parâmetro | Classe III | Classe IV |
|---|---|---|
| Prazo estimado | 4 a 12 meses | 9 a 20 meses |
| Taxa ANVISA (TFVS) | R$ 8.510 a R$ 14.800 | R$ 14.800 a R$ 19.856 |
| CBPF estrangeiro | ~R$ 72.805 | ~R$ 72.805 |
| Validade do registro | 10 anos | 10 anos |
Para mais detalhes sobre o processo passo a passo, consulte o artigo Como Registrar um Dispositivo Médico na ANVISA.
Via AREE: Análise Otimizada para Produtos Aprovados Internacionalmente
A Via AREE (Avaliação de Registro em Economias Equivalentes)
Os mercados reconhecidos como economias equivalentes são:
- FDA (Estados Unidos), para produtos com 510(k) ou PMA aprovados
- Health Canada (Canadá)
- TGA (Austrália)
- MHLW (Japão)
A principal vantagem da Via AREE é a redução de aproximadamente 30% no tempo de análise
Para fabricantes com aprovação prévia em um desses mercados, a Via AREE deve ser considerada prioritariamente na estratégia regulatória brasileira.
Boas Práticas de Fabricação: CBPF e MDSAP
O Certificado de Boas Práticas de Fabricação (CBPF) é obrigatório para dispositivos Classe III e IV. Ele atesta que o fabricante opera conforme os requisitos da RDC 665/2022ISO 13485.
Existem duas vias para obtenção do CBPF:

Via Direta, Inspeção ANVISA
O fabricante solicita inspeção à ANVISA, que realiza auditoria presencial no local de fabricação. O CBPF emitido tem validade de 2 anos. Essa via é obrigatória para fabricantes nacionais e opcional para estrangeiros.
Via MDSAP, Medical Device Single Audit Program
O MDSAP é um programa de auditoria única reconhecido por cinco países: Brasil, Estados Unidos, Canadá, Austrália e Japão. A auditoria é realizada por Organizações Auditoras (OAs) credenciadas pelo programa, e o relatório resultante é aceito pela ANVISA como base para emissão do CBPF, com validade ampliada para 4 anos (conforme RDC 850/2024).
A adoção do MDSAP por fabricantes que operam no Brasil cresceu de forma expressiva: de apenas 4,7% em 2017 para 59,1% em 2023
Existiam 87.758 licencas ativas de dispositivos medicos no Brasil em 31 de dezembro de 2023. Em 2023, foram emitidas 7.891 novas autorizacoes: 2.711 de Classe I, 4.162 de Classe II, 718 de Classe III e 300 de Classe IV.
— ANVISA, apresentacao no forum IMDRF, marco de 2024
Adicionalmente, a RDC 982/2025 introduziu o uso de inteligência artificial nos processos de certificação de BPF, sinalizando a modernização contínua do sistema de vigilância de boas práticas.
SaMD: Software como Dispositivo Médico
A RDC 657/2022 estabeleceu o primeiro marco regulatório brasileiro dedicado exclusivamente a Software como Dispositivo Médico (SaMD). Essa regulamentação é fundamental para startups de healthtech, desenvolvedores de aplicativos clínicos e fornecedores de soluções de inteligência artificial em saúde.
A classificação de SaMD segue a Regra 11 da RDC 751/2022, com quatro níveis baseados na gravidade do dano potencial ao paciente e no impacto da informação gerada pelo software na decisão clínica:
| Classe SaMD | Situação Clínica | Significado da Informação | Exemplos |
|---|---|---|---|
| I | Não grave / rotineira | Informar o clínico | Apps de registro de sintomas, calculadoras de IMC |
| II | Grave ou rotineira | Auxiliar na decisão clínica | Software de triagem hospitalar, monitoramento remoto |
| III | Grave | Orientar o diagnóstico ou tratamento | Detecção de arritmias por IA, diagnóstico por imagem assistido |
| IV | Crítica ou grave | Decisão autônoma de tratamento | Software de dosagem automatizada em UTI, sistemas de cirurgia robótica |
O mercado brasileiro já conta com aproximadamente 500 softwares registrados como SaMDsoftware adaptávelPredetermined Change Control Plan (PCCP) e requisitos de cibersegurança baseados na IEC 81001-5-1.
Em novembro de 2025, a ANVISA aprovou um Plano de Modernizacao abrangente: reducao de 40% no backlog pos-registro, investimento de R$ 25 milhoes em inteligencia artificial (projeto AnvisAI) e contratacao de 102 novos especialistas, a maior expansao desde 2014.
— ANVISA, Plano de Modernizacao, novembro de 2025
Certificação INMETRO para Equipamentos Eletromédicos
Além do registro ANVISA, equipamentos eletromédicos estão sujeitos à certificação compulsória do INMETRO
O processo segue o Modelo 5
- Ensaios de laboratório acreditado conforme IEC 60601-1 (segurança básica de equipamentos eletromédicos)
- Auditorias periódicas do sistema de qualidade do fabricante
- Vigilância de mercado contínua
O certificado INMETRO tem validade de 5 anos e deve ser mantido em conjunto com o registro ANVISA para comercialização legal no território nacional. Fabricantes devem planejar essas duas certificações de forma integrada, pois os ensaios exigidos frequentemente se sobrepõem.
Tecnovigilância e Pós-Mercado
O sistema de tecnovigilância brasileiro é operado por meio de duas plataformas principais: o NOTIVISA (Sistema de Notificações em Vigilância Sanitária) e o SISTEC (Sistema de Informações e Notificações em Tecnovigilância). Juntos, esses sistemas registram mais de 5.060 alertas ativos de dispositivos médicos.
Os prazos para notificação de eventos adversos são determinados pela gravidade:
| Tipo de Evento | Prazo de Notificação |
|---|---|
| Óbito do paciente | 72 horas |
| Evento grave (hospitalização, sequela) | 10 dias corridos |
| Mau funcionamento sem dano grave | 30 dias corridos |
O não cumprimento dos prazos de notificação pode resultar em sanções administrativas, incluindo advertências, multas e suspensão do registro do produto. A estruturação de um processo interno robusto de vigilância pós-comercialização é, portanto, parte indissociável da estratégia regulatória.
O setor de dispositivos medicos como um todo movimentou R$ 26,1 bilhoes em producao industrial em 2024, gerando 85.078 empregos diretos. Segundo o Termometro ABIMED 2025, 48% das empresas projetam crescimento superior a 10% e 61% planejam investir em inteligencia artificial.
— ABIMO, Relatorio Setorial 2025; ABIMED, Termometro 2025
Normas Técnicas Aplicáveis
O cumprimento das normas técnicas internacionais é a base para demonstração de conformidade com os Requisitos Essenciais de Segurança e Desempenho (RESD) da RDC 848/2024. As principais normas aplicáveis ao setor são:
| Norma | Escopo |
|---|---|
| IEC 60601-1 | Segurança básica e desempenho essencial de equipamentos eletromédicos |
| ISO 13485 | Sistemas de gestão da qualidade para dispositivos médicos |
| ISO 14971 | Gestão de riscos para dispositivos médicos |
| IEC 62304 | Ciclo de vida de software para dispositivos médicos |
| IEC 62366-1 | Engenharia de usabilidade para dispositivos médicos |
| IEC 81001-5-1 | Cibersegurança para software de saúde e dispositivos médicos |
A cibersegurança merece destaque especial: a RDC 848/2024 incluiu requisitos explícitos de proteção contra ameaças cibernéticas, especialmente relevantes para dispositivos conectados (IoMT, Internet of Medical Things) e SaMDs com comunicação de rede.
Brasil no Cenário Regulatório Internacional
O Brasil é membro fundador do IMDRF (International Medical Device Regulators Forum) desde 2011, e participa do MDSAP junto a EUA, Canadá, Austrália e Japão. Essa posição confere ao país um papel ativo na harmonização regulatória global.
Uma comparação objetiva com outros mercados relevantes ajuda a dimensionar a competitividade regulatória brasileira para o acesso ao mercado brasileiro:
| Critério | ANVISA (Brasil) | FDA (EUA) | EU MDR (Europa) |
|---|---|---|---|
| Custo máximo para alto risco | ~R$ 90 mil | ~US$ 350 mil (PMA) | € 50-150 mil (ON) |
| Prazo para alto risco | 9 a 20 meses | 12 a 36 meses | 12 a 24 meses |
| Reconhecimento mútuo | Via AREE (4 mercados) | Limitado | Organismos notificados |
| Alinhamento às normas ISO/IEC | Alto (pós-2022) | Parcial | Total (EU MDR 2017/745) |
Para fabricantes com aprovação FDA, a combinação da Via AREE com o MDSAP representa o caminho mais eficiente para o mercado brasileiro, reduzindo tanto o tempo quanto os custos regulatórios. Um exemplo concreto desse processo pode ser conferido no nosso estudo sobre a aprovação FDA para empresas internacionais, que detalha os desafios e estratégias para obter a autorização americana.
Tendências e Inovações Regulatórias para 2026 e Além
O ambiente regulatório brasileiro para dispositivos médicos segue em evolução acelerada. As principais tendências a monitorar são:
- UDI/SIUD: o cronograma de implantação do Sistema de Identificação Única de Dispositivos está em vigor com datas por classe entre 2025 e 2028, seguindo o modelo do UDI europeu e americano. Veja também como a ANVISA aprovou o sistema de rastreamento e identificação de dispositivos
- Impressão 3D: regulamentação específica em desenvolvimento para dispositivos fabricados por manufatura aditiva, com foco em implantes personalizados
- Sandbox Regulatório ANVISA: programa de ambiente controlado para inovações tecnológicas, permitindo testes com supervisão regulatória antes da submissão formal
- Marco Legal da IA (PL 2.338/2023): em tramitação no Congresso, com impacto direto sobre SaMDs com inteligência artificial e responsabilidade civil
- IoMT e cibersegurança: exigências crescentes da RDC 848/2024 para dispositivos conectados à internet ou a redes hospitalares
Fabricantes e startups que anteciparem esses requisitos em suas estratégias de desenvolvimento de produto terão vantagem competitiva significativa no momento da submissão regulatória.
Perguntas Frequentes
Qual é a diferença entre notificação e registro na ANVISA para dispositivos médicos?
Notificação é o processo aplicável a dispositivos de Classe I e II (risco baixo e médio). É mais simples, mais rápido (cerca de 30 dias) e tem custo menor (aproximadamente R$ 1.406). Registro é exigido para dispositivos de Classe III e IV (alto e máximo risco), exige CBPF obrigatório, dossiê técnico completo, e tem prazos de 4 a 20 meses e custos significativamente maiores. O registro tem validade de 10 anos, enquanto a notificação é por prazo indefinido.
O que é o CBPF e quem é obrigado a tê-lo?
O Certificado de Boas Práticas de Fabricação (CBPF) é o documento que atesta que o fabricante opera conforme os requisitos da RDC 665/2022 e da ISO 13485. É obrigatório para todos os fabricantes, de dispositivos médicos Classe III e IV. Pode ser obtido por inspeção direta da ANVISA (validade 2 anos) ou por auditoria MDSAP reconhecida (validade 4 anos, conforme RDC 850/2024).
Um aplicativo de saúde precisa de registro na ANVISA?
Depende da finalidade do software. Se o aplicativo for utilizado para diagnóstico, tratamento, monitoramento ou prevenção de doenças, ele provavelmente se enquadra na definição de SaMD da RDC 657/2022 e precisa de registro ou notificação conforme sua classe de risco (determinada pela Regra 11 da RDC 751/2022). Apps puramente educacionais ou de bem-estar sem finalidade diagnóstica ou terapêutica geralmente ficam fora do escopo regulatório. Em caso de dúvida, a consulta prévia à ANVISA é recomendada.
Como a Via AREE pode acelerar o registro no Brasil para fabricantes estrangeiros?
A Via AREE (RDC 741/2022, atualizada pela IN 290/2024) permite que fabricantes com aprovação prévia pela FDA, Health Canada, TGA ou MHLW utilizem a avaliação técnica daquelas agências como base para o registro brasileiro, reduzindo aproximadamente 30% do tempo de análise pela ANVISA. O fabricante ainda precisa apresentar documentação complementar (rotulagem em português, dados de vigilância pós-mercado locais), mas evita a análise técnica completa do zero.
Quais são os prazos obrigatórios para notificar eventos adversos à ANVISA?
Os prazos são determinados pela gravidade do evento: óbito deve ser notificado em até 72 horas; eventos graves (hospitalização não prevista, sequelas permanentes) em até 10 dias corridos; e mau funcionamento sem dano grave em até 30 dias corridos. O canal oficial para essas notificações é o sistema NOTIVISA da ANVISA. Fabricantes e importadores são igualmente responsáveis pelas notificações de tecnovigilância.
Este guia é atualizado periodicamente para refletir as mudanças no marco regulatório da ANVISA. Para aprofundamento em temas específicos, consulte os artigos relacionados: RDC 751/2022, Boas Práticas de Fabricação e SaMD, Dispositivos MédicosABIMO, International Medical Device Regulators Forum.
Conteúdo elaborado pela equipe editorial do Guia Definitivo de Engenharia Biomédica
Artigos relacionados
Certificação INMETRO para Equipamentos Médicos: Processo Passo a Passo
Guia completo sobre a certificação INMETRO de equipamentos eletromédicos no Brasil: Portaria 384/2020, normas IEC 60601, laboratórios acreditados, custos, prazos e o processo detalhado passo a passo para fabricantes e importadores.
Como Registrar um Dispositivo Médico na ANVISA: Roteiro Prático para Startups e Fabricantes
Guia completo do processo de registro de dispositivos médicos na ANVISA: classificação, CBPF, ensaios técnicos, submissão no sistema SOLICITA e acompanhamento. Prazos reais, custos atualizados e erros mais comuns a evitar.
IEC 60601: A Norma de Segurança de Equipamentos Eletromédicos Que Todo Engenheiro Precisa Conhecer
Entenda a estrutura completa da IEC 60601, o modelo sanduíche de normas gerais, colaterais e particulares, os conceitos de MOPP/MOOP, classificação de partes aplicadas e os requisitos de ensaio que definem a segurança de equipamentos eletromédicos no Brasil e no mundo.
ISO 13485 para Dispositivos Médicos: Implementação Prática do Sistema de Gestão da Qualidade
Guia completo de implementação da ISO 13485:2016 com checklists por cláusula, templates de procedimentos, custos reais de certificação e estratégias para auditoria. Essencial para gestores de qualidade e consultores do setor de dispositivos médicos no Brasil.