A RDC 665/2022 é o marco regulatório central das Boas Práticas de Fabricação (BPF) de produtos médicos no Brasil. Publicada pela ANVISA em 30 de março de 2022 e vigente desde 2 de maio de 2022, ela consolidou e substituiu a RDC 16/2013 e a IN 8/2013, reunindo em um único instrumento todos os requisitos que fabricantes, importadores, distribuidores e armazenadores de dispositivos médicos devem cumprir para operar legalmente no país. Seu escopo abrange 138 artigos e incorpora a Resolução GMC 20/2011 do Mercosul, harmonizando o Brasil com o padrão regional e aproximando o arcabouço regulatório nacional da ISO 13485:2016.
Se você atua na gestão de qualidade ou na fabricação de dispositivos médicos, este artigo detalha cada pilar da norma, os documentos obrigatórios, os caminhos para obtenção do Certificado de Boas Práticas de Fabricação (CBPF) e um checklist prático de conformidade. Para contextualizar a RDC 665/2022 dentro do ecossistema regulatório mais amplo, consulte também nosso Guia Completo ANVISA para Dispositivos Médicos 2026.
O que Mudou com a RDC 665/2022 em Relação à RDC 16/2013
A primeira informação crítica para gestores é: a RDC 665/2022 não introduziu requisitos substantivos novos em comparação com a RDC 16/2013. Seu principal propósito foi consolidação e reestruturação. Isso significa que empresas já em conformidade com a norma anterior não precisaram reformular seus sistemas de gestão da qualidade, o que foi necessário foi revisar a numeração dos artigos e atualizar as referências internas na documentação.
As principais diferenças formais incluem:
- Incorporação expressa da Resolução GMC 20/2011 do Mercosul, explicitando o alinhamento regional;
- Unificação do texto normativo anteriormente dividido entre a RDC 16/2013 e a IN 8/2013;
- Reestruturação da numeração dos artigos para maior clareza e navegabilidade;
- Atualização de definições para alinhamento com a ISO 13485:2016 e o MDSAP (Medical Device Single Audit Program).
Para entender como a RDC 665/2022 se encaixa no processo de registro de produtos, veja nosso artigo sobre a RDC 751/2022: Classificação e Registro de Dispositivos Médicos.
Os 138 Artigos: Estrutura e Pilares da RDC 665/2022
A norma está organizada em blocos temáticos que cobrem o ciclo de vida completo do dispositivo médico. A tabela abaixo apresenta os principais blocos e seus temas centrais:
| Bloco Temático | Temas Principais | Relevância para SGQ |
|---|---|---|
| Sistema de Gestão da Qualidade | Estrutura do SGQ, planejamento, controle de documentos e registros | Base de todo o sistema; alinhado à ISO 13485 |
| Responsabilidade da Direção | Comprometimento, política da qualidade, planejamento, autoridade e comunicação | Alta gestão deve demonstrar liderança ativa |
| Gestão de Recursos | Recursos humanos, competência, infraestrutura e ambiente de trabalho | Treinamentos documentados e qualificação de pessoal |
| Gestão de Riscos | Análise, avaliação, controle e verificação de eficácia ao longo do ciclo de vida | Processo contínuo; integrado ao projeto e produção |
| Controles de Projeto e Desenvolvimento | Planejamento, entradas, saídas, revisões, verificação, validação, transferência e mudanças | Gate reviews e Registro Histórico do Projeto |
| Controles de Compras | Qualificação e avaliação de fornecedores, requisitos de compra, verificação | Lista de fornecedores aprovados e auditorias |
| Controles de Produção | Ambiente, equipamentos, validação de processos, identificação, rastreabilidade | Validação obrigatória onde outputs não são verificáveis |
| Manuseio, Armazenamento e Distribuição | Rotulagem, embalagem, controle de estoque, distribuição e rastreabilidade | Cadeia de custódia e controle de lote |
| Instalação e Serviço | Registros de instalação, serviço pós-venda, relatórios de serviço | Aplicável quando contratualmente acordado |
| CAPA e Reclamações | Tratamento de reclamações, investigação, ações corretivas/preventivas, recalls | Ciclo fechado com verificação de eficácia |
| Técnicas Estatísticas | Amostragem, controle estatístico de processo, análise de dados | Procedimentos documentados obrigatórios |
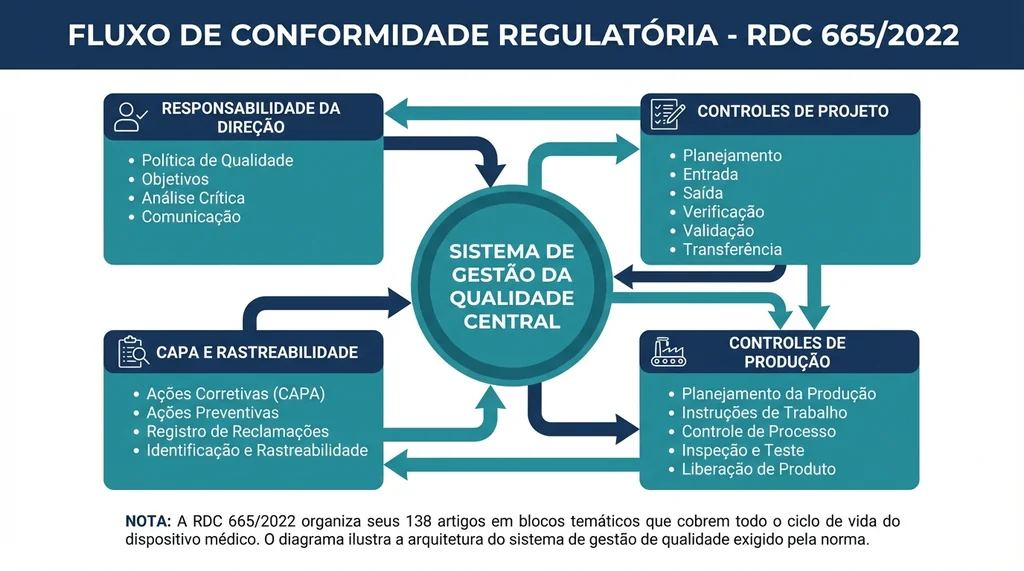
Descrição completa da imagem
Diagrama técnico mostrando a estrutura dos 138 artigos da RDC 665/2022 e os pilares do sistema de gestão da qualidade para dispositivos médicos.
Documentos Obrigatórios: RMP, RHP e Registro Histórico do Projeto
Um dos pontos mais fiscalizados em auditorias é a existência e integridade dos três documentos mestres exigidos pela RDC 665/2022. Compreender a distinção entre eles é fundamental:
Registro Mestre do Produto (RMP)
O RMP é o conjunto de documentos que define como o produto deve ser fabricado. Ele contém as especificações do produto, procedimentos de fabricação, equipamentos necessários, métodos de inspeção e ensaio, e requisitos de embalagem e rotulagem. É o "como fazer" do produto, e deve estar disponível para todos os envolvidos na produção.
Registro Histórico do Produto (RHP)
O RHP é o conjunto de registros que demonstra que cada lote ou unidade foi fabricado em conformidade com o RMP. Inclui registros de produção, resultados de ensaios, dados de rastreabilidade de componentes, desvios documentados e ações tomadas. É o "como foi feito", a evidência objetiva de conformidade por lote.
Registro Histórico do Projeto
Exclusivo para dispositivos com controles de projeto aplicáveis, este documento registra todo o processo de desenvolvimento: planejamento, entradas de projeto, revisões realizadas, resultados de verificação e validação, e mudanças de projeto com respectivas aprovações. É a trilha de auditoria do ciclo de desenvolvimento do produto.
Atenção para auditorias:
Auditores ANVISA e MDSAP verificam sistematicamente a rastreabilidade entre RMP, RHP e Registro Histórico do Projeto. Inconsistências entre esses documentos são uma das não conformidades mais frequentes em inspeções.
Gestão de Riscos: Processo Contínuo ao Longo do Ciclo de Vida
A RDC 665/2022 exige que a gestão de riscos seja um processo contínuo e não um evento pontual. Isso alinha o Brasil com os requisitos da ISO 14971 e com o entendimento internacional de que riscos devem ser monitorados durante toda a vida útil do produto no mercado.
O processo completo de gestão de riscos exigido pela norma compreende quatro etapas integradas:
| Etapa | Atividades Requeridas | Integração com outros requisitos |
|---|---|---|
| Análise de Riscos | Identificação de hazards, estimativa de probabilidade e severidade | Entradas de projeto, especificações de uso pretendido |
| Avaliação de Riscos | Decisão sobre aceitabilidade com base em critérios pré-definidos | Política da qualidade, política de risco da empresa |
| Controle de Riscos | Implementação de medidas: design seguro, proteções, informações de segurança | Controles de projeto, rotulagem, IFU |
| Verificação de Eficácia | Confirmação de que controles reduziram riscos ao nível aceitável sem introduzir novos riscos | Dados pós-mercado, reclamações, CAPA |
A integração da gestão de riscos com dados pós-mercado é particularmente importante: reclamações e eventos adversos devem alimentar reanálises de risco, fechando o ciclo de melhoria contínua. Para dispositivos que utilizam inteligência artificial, a RDC 982/2025 estabeleceu diretrizes adicionais para a incorporação de IA na gestão de riscos dentro do contexto BPF.
A RDC 665/2022 exige que cada fabricante estabeleça e mantenha um processo contínuo de gerenciamento de risco que envolva todo o ciclo de vida do produto, incluindo análise de risco, avaliação de risco, controle de risco e avaliação da efetividade dos controles estabelecidos.
— ANVISA, RDC nº 665/2022, Art. 18–19
A RDC 665/2022 incorpora ao ordenamento jurídico brasileiro a Resolução GMC MERCOSUL nº 20/2011, harmonizando os requisitos de BPF com o padrão regional e facilitando o comércio de dispositivos médicos entre os países membros do bloco.
— ANVISA, Diário Oficial da União, 31 de março de 2022
Controles de Produção e Validação de Processos
A RDC 665/2022 estabelece um princípio fundamental para controles de produção: qualquer processo cujo output não possa ser completamente verificado por inspeção ou ensaio subsequente deve ser validado. Isso inclui processos como esterilização, soldagem, selagem de embalagens, tratamentos superficiais e aplicação de revestimentos bioativos.
A validação de processos segue a lógica IQ/OQ/PQ (Qualificação de Instalação, Operação e Performance), devendo ser documentada antes do início da produção comercial. Mudanças em equipamentos, instalações ou processos validados exigem reavaliação do impacto e eventual revalidação.
Além da validação, a norma exige:
- Identificação e rastreabilidade: cada componente, subconjunto e produto acabado deve ser identificável ao longo de todas as etapas de produção;
- Controle de produto não conforme: procedimentos documentados para identificar, segregar, avaliar e dispor de produtos que não atendam às especificações;
- Controle ambiental: monitoramento e controle das condições ambientais de produção quando relevantes para a qualidade do produto (temperatura, umidade, partículas, etc.);
- Calibração e manutenção de equipamentos: programa documentado de calibração e manutenção preventiva para todos os equipamentos de medição e produção críticos.

CBPF: Certificado de Boas Práticas de Fabricação
O Certificado de Boas Práticas de Fabricação (CBPF) é obrigatório para o registro de dispositivos médicos das Classes III e IV, as de maior risco, junto à ANVISA. Sem o CBPF válido, o processo de registro não avança para estas classes.
A RDC 665/2022, combinada com as diretrizes subsequentes, prevê dois caminhos para obtenção do CBPF:
| Caminho | Mecanismo | Validade do CBPF | Principais Vantagens |
|---|---|---|---|
| Inspeção Direta ANVISA | Inspeção presencial realizada por auditores da ANVISA na instalação do fabricante | 2 anos | Não requer certificação por organismo externo; adequado para empresas nacionais menores |
| Relatórios MDSAP (RDC 850/2024) | Submissão de relatórios de auditoria MDSAP realizados por Organismo Auditor acreditado | 4 anos | Validade maior; auditoria única reconhecida por múltiplas agências regulatórias internacionais |
MDSAP e ISO 13485: Alinhamento Internacional
O MDSAP (Medical Device Single Audit Program) representa uma das mais significativas evoluções no cenário regulatório de dispositivos médicos dos últimos anos. O programa permite que uma única auditoria, realizada por um Organismo Auditor acreditado, satisfaça os requisitos de auditoria de múltiplas agências regulatórias, incluindo ANVISA (Brasil), FDA (EUA), Health Canada, TGA (Austrália) e PMDA (Japão).
A adoção do MDSAP no Brasil cresceu de forma expressiva: de apenas 4,7% das empresas participantes em 2017 para 59,1% em 2023. Em 2025, existem 13 Organismos Auditores acreditados para conduzir auditorias MDSAP globalmente.
A relação entre MDSAP e ISO 13485:2016 é de complementaridade: as auditorias MDSAP avaliam a conformidade do SGQ com a ISO 13485:2016 ao mesmo tempo em que verificam os requisitos regulatórios específicos de cada país participante. Para empresas que buscam certificação ISO 13485 para Dispositivos Médicos a auditoria MDSAP oferece dupla cobertura, eficiência operacional e conformidade regulatória simultânea.
Benefício estratégico do MDSAP:
Empresas com produto aprovado no Brasil que buscam expandir para o mercado norte-americano ou canadense encontram no MDSAP um atalho regulatório significativo, evitando auditorias redundantes por diferentes agências.
A RDC 850/2024 ampliou a validade do Certificado de Boas Práticas de Fabricação de dois para quatro anos para fabricantes participantes do programa MDSAP, em contrapartida a auditorias anuais de monitoramento.
— ANVISA, RDC nº 850/2024
Checklist de Conformidade com a RDC 665/2022
A implementação prática da RDC 665/2022 exige uma abordagem sistemática. O checklist abaixo organiza os requisitos essenciais por área, permitindo identificar gaps no sistema de gestão da qualidade:
| Área | Requisito Essencial | Evidência Objetiva Necessária |
|---|---|---|
| SGQ | Manual da qualidade ou documento equivalente definindo escopo e exclusões justificadas | Manual aprovado pela direção, com data e versão |
| Direção | Política da qualidade, objetivos mensuráveis e análise crítica pela direção | Atas de análise crítica com frequência mínima anual |
| Pessoal | Matrizes de competência, treinamentos documentados e avaliação de eficácia | Registros de treinamento assinados e avaliações de eficácia |
| Gestão de Riscos | Arquivo de gestão de riscos por produto, cobrindo todo o ciclo de vida | Plano de gestão de riscos, análise FMEA, relatório de gestão de riscos |
| Projeto | Plano de projeto, revisões formais em gates, verificação e validação de projeto | Registro Histórico do Projeto completo e rastreável |
| Fornecedores | Critérios de qualificação, lista de fornecedores aprovados, avaliação periódica | AVF aprovados, registros de avaliação anual ou por risco |
| Produção | Validação de processos especiais, controle ambiental, rastreabilidade de lote | Relatórios IQ/OQ/PQ, registros de lote (RHP), dados de monitoramento ambiental |
| Calibração | Inventário de instrumentos, cronograma de calibração, rastreabilidade metrológica | Certificados de calibração rastreáveis ao INMETRO ou equivalente |
| CAPA | Processo documentado para reclamações, NC, CAPA e verificação de eficácia | Log de CAPA com status, prazos e registros de fechamento com eficácia verificada |
| Auditoria Interna | Programa anual de auditorias internas cobrindo todos os requisitos da norma | Relatórios de auditoria, planos de ação e evidências de closure |
Para equipamentos que também requerem certificação do INMETRO, consulte nosso artigo sobre Certificação INMETRO para Equipamentos Médicos que detalha como a conformidade BPF complementa os requisitos de certificação de produto.
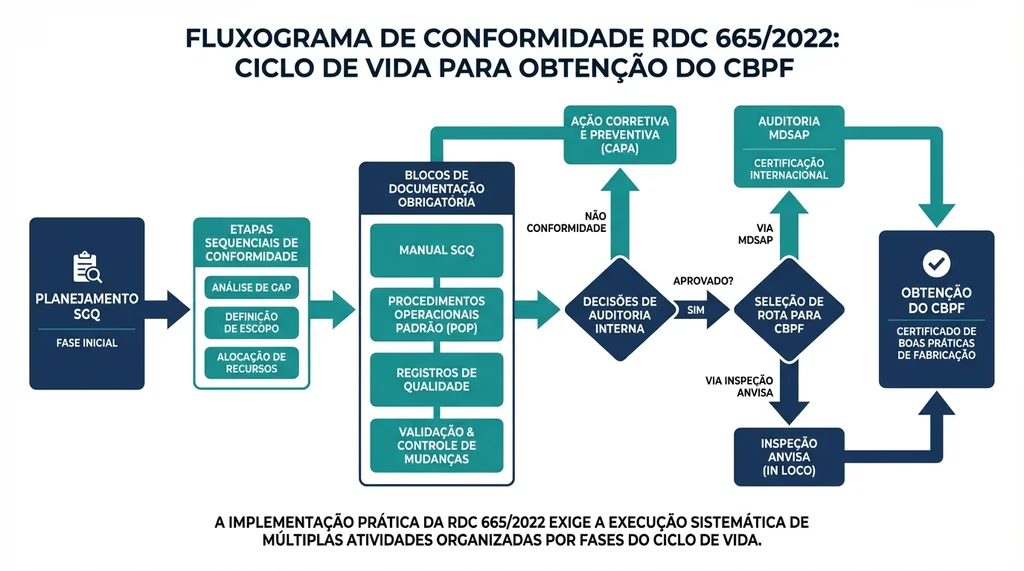
Descrição completa da imagem
Fluxograma do processo de conformidade com a RDC 665/2022 desde implementação do SGQ até obtenção do CBPF.
CAPA: O Motor da Melhoria Contínua
O sistema de Ações Corretivas e Preventivas (CAPA) é frequentemente descrito como o coração de qualquer SGQ eficaz, e a RDC 665/2022 não é exceção. A norma exige um processo CAPA robusto que abranja:
- Reclamações de clientes: todas as reclamações devem ser registradas e avaliadas quanto ao potencial de representar um evento adverso reportável à ANVISA;
- Investigação de causas raízes: uso de metodologias como 5-Why, Ishikawa (espinha de peixe) ou análise de árvore de falhas para identificar a causa fundamental do problema;
- Ações corretivas: eliminação da causa de não conformidades existentes para prevenir reincidência;
- Ações preventivas: eliminação de causas de não conformidades potenciais antes que ocorram;
- Recalls: processo documentado para identificação de lotes afetados, notificação à ANVISA e recolhimento de produtos quando necessário.
Um requisito frequentemente negligenciado é a verificação formal de eficácia das CAPA: após implementação da ação, a empresa deve demonstrar objetivamente que o problema foi resolvido e que a solução se mantém ao longo do tempo. CAPA sem verificação de eficácia é uma das não conformidades mais recorrentes em inspeções ANVISA.
RDC 982/2025: Inteligência Artificial e BPF
Em 2025, a ANVISA publicou a RDC 982/2025, que introduziu diretrizes para a integração de inteligência artificial nos processos de gestão de riscos dentro do contexto das BPF. Esta norma é particularmente relevante para fabricantes que utilizam IA em sistemas de visão computacional para controle de qualidade, monitoramento preditivo de processos ou análise de dados de vigilância pós-mercado.
As exigências incluem validação dos algoritmos de IA, documentação de seus limites de performance, procedimentos para atualização de modelos e monitoramento contínuo de drift de performance. Embora complementar à RDC 665/2022, a RDC 982/2025 representa o próximo horizonte regulatório para empresas que incorporam tecnologias de IA em seus processos produtivos e de qualidade.
Perguntas Frequentes sobre a RDC 665/2022
A RDC 665/2022 se aplica a distribuidores e importadores, ou apenas a fabricantes?
A norma aplica-se a fabricantes, importadores, distribuidores e armazenadores de dispositivos médicos que atuam no mercado brasileiro. Os requisitos específicos variam conforme o papel da empresa na cadeia: fabricantes têm o maior escopo de requisitos aplicáveis, incluindo controles de projeto e produção. Importadores e distribuidores devem cumprir principalmente os requisitos de manuseio, armazenamento, distribuição, controle de documentos, CAPA e rastreabilidade.
O CBPF é obrigatório para dispositivos médicos de Classe I e II?
Não. O CBPF é exigência para o registro de dispositivos médicos das Classes III e IV. Para Classes I e II, a empresa deve igualmente cumprir com os requisitos de BPF da RDC 665/2022, mas não é necessário apresentar o CBPF como condição de registro. No entanto, estar em conformidade com as BPF é obrigatório independentemente da classe, e a ANVISA pode realizar inspeções em qualquer empresa fabricante, independentemente do porte ou classe dos produtos.
Qual a diferença entre o caminho MDSAP e a inspeção direta ANVISA para o CBPF?
Os dois caminhos levam ao mesmo resultado, o CBPF, mas diferem em validade e processo. A inspeção direta pela ANVISA resulta em CBPF com validade de 2 anos; o caminho via relatórios MDSAP, regulamentado pela RDC 850/2024, resulta em CBPF com validade de 4 anos. O MDSAP é especialmente vantajoso para empresas que exportam para múltiplos países participantes (EUA, Canadá, Austrália, Japão), pois uma única auditoria satisfaz os requisitos regulatórios de todos eles simultaneamente.
Como a validação de processos deve ser documentada segundo a RDC 665/2022?
A norma exige que a validação de processos especiais seja realizada e documentada antes do início da produção comercial. A documentação deve incluir: protocolo de validação com critérios de aceitação pré-definidos, resultados das qualificações IQ (Instalação), OQ (Operação) e PQ (Performance), relatório de validação com conclusão formal sobre aprovação ou rejeição, e programa de revalidação periódica ou por mudança. Mudanças nos equipamentos, instalações, pessoal ou parâmetros de processo devem ser avaliadas quanto ao impacto e podem exigir revalidação total ou parcial.
Uma empresa certificada na ISO 13485:2016 já está em conformidade com a RDC 665/2022?
A ISO 13485:2016 e a RDC 665/2022 têm alto grau de alinhamento, e uma empresa certificada na norma internacional certamente já atende grande parte dos requisitos da norma brasileira. No entanto, existem requisitos regulatórios específicos da ANVISA que vão além da ISO 13485, como exigências de rotulagem para o mercado brasileiro, procedimentos de notificação de eventos adversos à ANVISA, requisitos específicos do Mercosul e particularidades do CBPF. Portanto, a certificação ISO 13485 é uma excelente base, mas não substitui completamente a análise de conformidade com a RDC 665/2022.
Fontes e Referências Normativas
Artigos relacionados
ANVISA e Dispositivos Médicos: Guia Completo de Regulamentação [Atualizado 2026]
Guia completo sobre regulamentação de dispositivos médicos pela ANVISA: classes de risco I-IV, fluxo de registro, documentação exigida, prazos, custos e as principais RDCs de 2022 a 2025. Conteúdo atualizado para profissionais de regulatory affairs, startups e fabricantes.
Certificação INMETRO para Equipamentos Médicos: Processo Passo a Passo
Guia completo sobre a certificação INMETRO de equipamentos eletromédicos no Brasil: Portaria 384/2020, normas IEC 60601, laboratórios acreditados, custos, prazos e o processo detalhado passo a passo para fabricantes e importadores.
Como Registrar um Dispositivo Médico na ANVISA: Roteiro Prático para Startups e Fabricantes
Guia completo do processo de registro de dispositivos médicos na ANVISA: classificação, CBPF, ensaios técnicos, submissão no sistema SOLICITA e acompanhamento. Prazos reais, custos atualizados e erros mais comuns a evitar.
IEC 60601: A Norma de Segurança de Equipamentos Eletromédicos Que Todo Engenheiro Precisa Conhecer
Entenda a estrutura completa da IEC 60601, o modelo sanduíche de normas gerais, colaterais e particulares, os conceitos de MOPP/MOOP, classificação de partes aplicadas e os requisitos de ensaio que definem a segurança de equipamentos eletromédicos no Brasil e no mundo.