A RDC 751/2022 é a principal norma regulatória de dispositivos médicos no Brasil substituindo a RDC 185/2001 e consolidando mais de duas décadas de evolução regulatória em um único instrumento legal. Publicada em 21 de setembro de 2022 e em vigor desde 1º de março de 2023, ela define como fabricantes, importadores e distribuidores devem classificar, notificar e registrar produtos junto à ANVISA, impactando diretamente os profissionais de regulatory affairs e engenheiros de qualidade que atuam nesse setor.
Este artigo faz parte do Guia Definitivo de Engenharia Biomédica.
Contexto Histórico e Revogações
A RDC 751/2022 não surgiu no vácuo. Ela consolidou e revogou seis normas anteriores que coexistiam de forma fragmentada, gerando insegurança jurídica para as empresas. Entender o que foi revogado é fundamental para evitar referencias a normas obsoletas em documentos técnicos:
| Norma Revogada | Assunto | Substituída por |
|---|---|---|
| RDC 185/2001 | Registro de dispositivos médicos (norma mãe anterior) | RDC 751/2022 integral |
| RE 1.554/2002 | Lista de produtos dispensados de registro | Anexo I da RDC 751/2022 |
| RDC 207/2006 | Dispositivos de uso único | Seção específica da RDC 751/2022 |
| IN 4/2012 (itens específicos) | Instruções sobre dossiê técnico | IN 290/2024 + RDC 751/2022 |
| RDC 15/2014 | Regime de vigilância especial | Integrado às regras especiais (14-22) |
| RDC 40/2015 | Alterações pós-registro | Capítulo específico da RDC 751/2022 |
A harmonização com o arcabouço regulatório global foi outro objetivo central. As 22 regras de classificação da RDC 751/2022 espelham diretamente o Regulamento (UE) 2017/745 (EU MDR), facilitando a dupla submissão para empresas que atuam simultaneamente nos mercados europeu e brasileiro.
A RDC 751/2022 foi motivada pela internalizacao da Resolucao GMC n. 25/2021 do Mercosul, harmonizando requisitos entre Brasil, Argentina, Paraguai e Uruguai, e pelo alinhamento com o IMDRF, adotando sua estrutura de sumario para dossies tecnicos (IMDRF/RPS WG/N9 Edition 3).
— ANVISA, Nota Tecnica da RDC 751/2022; Mercosul, GMC 25/2021
As 4 Classes de Risco: Definições e Implicações Regulatórias
O sistema de quatro classes de risco é a espinha dorsal da RDC 751/2022. A classe determina a via de acesso ao mercado, os documentos exigidos, os prazos de análise e os custos envolvidos:
| Classe | Risco | Via | Prazo Legal | Validade | CBPF Exigido? | TFVS (base) |
|---|---|---|---|---|---|---|
| I | Baixo | Notificação | ~30 dias | Indefinida | Não | R$ 1.406 |
| II | Médio | Notificação | ~30 dias | Indefinida | Não | R$ 1.406 |
| III | Alto | Registro | 250–365 dias | 10 anos | Sim | R$ 8.510–14.000 |
| IV | Máximo | Registro | 250–365 dias | 10 anos | Sim | R$ 14.000–19.856 |
Vale destacar que o prazo mediano real para registro (classes III e IV) gira em torno de 389 dias, superando o prazo legal. Estratégias para reduzir esse tempo incluem a via AREE (Análise com Base em Evidências Regulatórias Estrangeiras), regulamentada pela IN 290/2024, que permite reliance em decisões do FDA, Health Canada, TGA (Austrália) e PMDA (Japão), reduzindo o tempo de análise em aproximadamente 30%.
Apenas 31% dos dispositivos medicos registrados no Brasil sao fabricados localmente, refletindo forte dependencia de importacoes que totalizaram US$ 9,79 bilhoes em 2024 (+20,49%). As exportacoes brasileiras atingiram US$ 1,17 bilhao (+24,6%), abrangendo mais de 190 paises.
— ABIMO, Relatorio Setorial 2025; ANVISA, dados de comercio exterior 2024
Para mais detalhes sobre o processo completo de registro, consulte nosso guia sobre como registrar dispositivo médico na ANVISA.
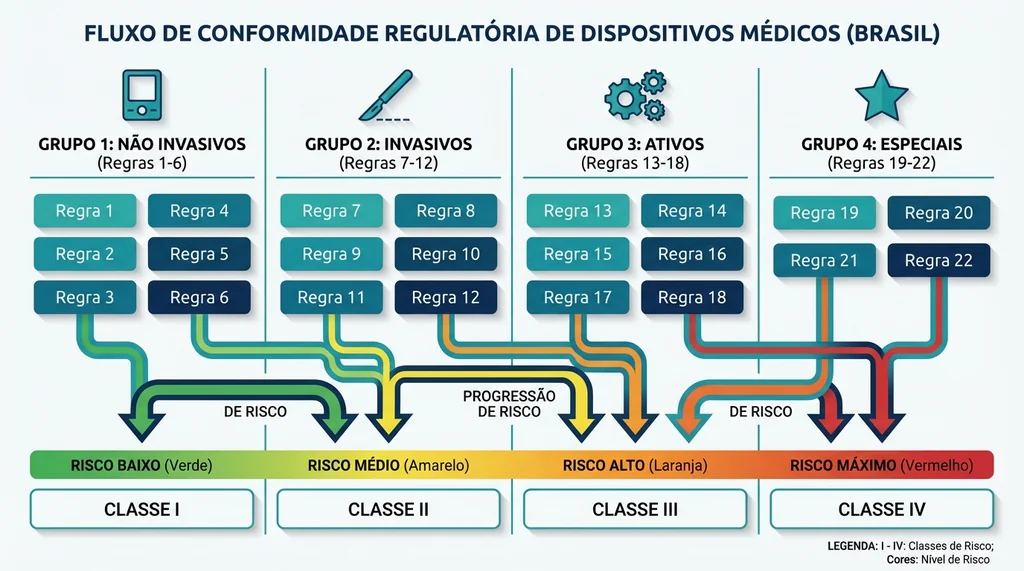
As 22 Regras de Classificação: Análise por Grupos
A classificação de um dispositivo médico é o passo mais crítico do processo regulatório. Errar a classe significa submeter documentação incorreta, podendo resultar em indeferimento ou, pior, irregularidade sanitária. A RDC 751/2022 organiza as 22 regras em quatro grupos:
Grupo 1, Dispositivos Não Invasivos (Regras 1 a 4)
As regras de 1 a 4 cobrem produtos que não penetram o corpo humano. A regra geral (Regra 1) classifica todos os não invasivos como Classe I, mas as regras subsequentes elevam a classe conforme o risco:

Descrição completa da imagem
Fluxograma das 22 regras de classificação RDC 751/2022 organizadas em 4 grupos de risco de dispositivos médicos ANVISA.
- Regra 1: Dispositivos não invasivos em geral, Classe I (ex.: termômetros de mercúrio, espátulas).
- Regra 2: Dispositivos para condução ou armazenamento de sangue, líquidos corporais ou tecidos, Classe II (ex.: bolsas coletoras de urina, tubos de drenagem).
- Regra 3: Dispositivos para modificar composição biológica ou química de sangue, Classe III (ex.: filtros de diálise).
- Regra 4: Dispositivos em contato com pele lesionada, Classe I (feridas superficiais) a Classe III (feridas crônicas, queimaduras de terceiro grau).
Grupo 2, Dispositivos Invasivos (Regras 5 a 8)
Invasivos são aqueles que penetram o corpo, parcial ou totalmente, por orifício corporal ou pela superfície corporal. A duração do contato (transitório, curto prazo, longo prazo) e o local de uso são os fatores determinantes:
- Regra 5: Invasivos por orifício corporal não cirúrgico, Classe I a III conforme duração e local (ex.: especulum vaginal = I; cateter uretral de longa duração = III).
- Regra 6: Invasivos cirúrgicos de uso transitório, Classe II (ex.: bisturis, luvas cirúrgicas); Classe III se reutilizáveis e para diagnóstico de doenças graves.
- Regra 7: Implantados de curto prazo, Classe II a III (ex.: drenos cirúrgicos absorvíveis).
- Regra 8: Implantados de longa duração, Classe III (ex.: parafusos ortopédicos de titânio); Classe IV se implantados no sistema nervoso central ou coração (ex.: marca-passo, válvulas cardíacas).
Grupo 3, Dispositivos Ativos (Regras 9 a 13)
Dispositivos ativos dependem de energia elétrica, eletromagnética ou outra fonte que não a força humana ou gravitacional. Este grupo inclui equipamentos de diagnóstico por imagem, ventiladores e desfibriladores:
- Regra 9: Dispositivos ativos terapêuticos, Classe I (ex.: aparelhos auditivos de amplificação simples); Classe IIb/III se risco em caso de falha (ex.: ventiladores mecânicos).
- Regra 10: Dispositivos ativos de diagnóstico, Classe I a II (ex.: eletrocardiógrafo = II); Classe III se fornecem energia ionizante (ex.: tomógrafo).
- Regra 11 (SaMD): Regra nova introduzida pela RDC 751/2022. Softwares como Dispositivos Médicos (SaMD) são classificados conforme o impacto clínico da informação gerada. Para saber mais, consulte nosso artigo sobre SaMD, Software como Dispositivo Médico.
- Regra 12: Dispositivos que administram medicamentos ou outros produtos, Classe II a III (ex.: bomba de infusão = III).
- Regra 13: Outros dispositivos ativos, Classe I por padrão.
Grupo 4, Regras Especiais (Regras 14 a 22)
As regras especiais cobrem categorias com características únicas que justificam tratamento diferenciado. Quatro delas foram introduzidas pela RDC 751/2022 e não existiam na RDC 185/2001:
| Regra | Categoria | Classe Típica | Exemplo |
|---|---|---|---|
| 14 | Contraceptivos e DSTs | II–III | Preservativos (III), DIU (III) |
| 15 | Desinfecção de dispositivos | II–III | Glutaraldeído (III) |
| 16 | Radiação ionizante | II–IV | Fontes radioativas implantáveis (IV) |
| 17 | Derivados de sangue ou tecidos | III–IV | Substitutos ósseos (III) |
| 18 | Uso em diagnóstico in vitro | I–IV | Glicosímetros (II) |
| 19 | Nanomateriais (nova) | III–IV | Nanopartículas terapêuticas (IV) |
| 20 | Inalação (nova) | II–III | Nebulizadores (II), inaladores pressurizados (III) |
| 21 | Substâncias absorvidas (nova) | III–IV | Adesivos transdérmicos medicamentosos (III) |
| 22 | Terapêutico + diagnóstico (nova) | Classe mais alta | Cateter ablação com imageamento (IV) |
Princípio do pior caso: quando mais de uma regra for aplicável ao mesmo dispositivo, prevalece a que resultar na classificação mais elevada. Esse princípio é expresso no artigo 11 da RDC 751/2022 e deve ser documentado no dossiê técnico.
Dossiê Técnico: Estrutura IMDRF e Documentação Exigida
A estrutura do dossiê técnico da RDC 751/2022 adota o sumário IMDRF (International Medical Device Regulators Forum), alinhando o Brasil ao padrão global. Isso permite o reaproveitamento de documentos preparados para o FDA 510(k), FDA PMA ou marcação CE:
Módulos do Dossiê para Registro (Classes III e IV)
| Módulo | Conteúdo Principal | Normas de Referência |
|---|---|---|
| 1, Administrativo | Formulários, procurações, TFVS, comprovantes de CBPF | RDC 751/2022, Art. 25–32 |
| 2, Visão Geral | Descrição do dispositivo, finalidade prevista, usuário pretendido | IMDRF/GRRP WG/N47 |
| 3, Não Clínico | Testes de biocompatibilidade, esterilização, EMC, software | ISO 10993, IEC 60601, IEC 62304 |
| 4, Clínico | Avaliação clínica, dados de desempenho clínico, PMCF | ABNT NBR ISO 14155, IMDRF/SaMD WG |
| 5, Gestão de Risco | Arquivo de gestão de risco, FMEA, análise de árvore de falhas | ISO 14971:2019 |
| 6, Fabricação | Processo produtivo, controle de qualidade, CBPF ou equivalente | RDC 665/2022 ISO 13485 |
| 7, Rotulagem | Rótulo, IFU em português, símbolos ISO 15223 | RDC 751/2022, Art. 55–68 |
Para notificações (Classes I e II), o dossiê é simplificado: dispensa avaliação clínica completa e aceita autodeclaração de conformidade às normas técnicas. O sistema SOLICITA da ANVISA é a plataforma eletrônica para submissão.
Certificado de Boas Práticas de Fabricação (CBPF)
O CBPF é exigido para Classes III e IV. Para fabricantes estrangeiros, a ANVISA aceita:
- Certificado MDSAP (Medical Device Single Audit Program), A taxa de adoção saltou de 4,7% em 2017 para 59,1% em 2023 entre fabricantes importados, tornando-se a opção preferida.
- Certificado de BPF emitido pela autoridade sanitária do país de origem, desde que o país tenha acordo de reconhecimento mútuo com o Brasil.
- Certificado emitido pela própria ANVISA após inspeção internacional.
Para fabricantes nacionais, o CBPF é emitido pela ANVISA após inspeção na planta produtiva. O sistema de qualidade deve estar conforme a ISO 13485.
Os certificados CBPF baseados em relatorios MDSAP saltaram de 38 em 2017 para 659 em 2023 (59,1% do total), evidenciando a consolidacao do programa como via preferencial para certificacao de fabricacao no Brasil.
— ANVISA, dados de certificacao BPF 2017-2023; IMDRF MDSAP Report
Rotulagem e Instruções de Uso
A RDC 751/2022 dedica os artigos 55 a 68 exclusivamente à rotulagem, tornando-a uma das seções mais detalhadas da norma. Os requisitos centrais são:
- Idioma obrigatório: português do Brasil para todos os elementos textuais do rótulo e das instruções de uso (IFU).
- Símbolos: permitido o uso de símbolos ISO 15223-1, desde que acompanhados de legenda ou glossário.
- IFU digital: autorizada pela RDC 751/2022, substituindo o impresso, desde que o acesso seja garantido por URL ou QR Code no rótulo físico. Exceção: dispositivos implantáveis e de uso domiciliar devem manter IFU impressa.
- Uso único: dispositivos de uso único devem trazer o símbolo específico e a proibição expressa de reprocessamento, salvo quando o fabricante disponibiliza protocolo validado de reprocessamento.
UDI/SIUD: Identificação Única de Dispositivos
A identificação única de dispositivos (UDI, Unique Device Identification) foi introduzida no Brasil pelo SIUD (Sistema de Identificação Única de Dispositivos Médicos), com cronograma de implementação faseado por classe de risco:
| Classe | Prazo para Atribuição de UDI | Prazo para Inclusão no SIUD |
|---|---|---|
| IV | Janeiro de 2025 | Julho de 2025 |
| III | Janeiro de 2026 | Julho de 2026 |
| II | Janeiro de 2027 | Julho de 2027 |
| I | Janeiro de 2028 | Julho de 2028 |
O UDI é composto pelo Device Identifier (DI), que identifica o modelo, e pelo Production Identifier (PI), que identifica o lote, número de série e data de fabricação. Para mais informações sobre o panorama do setor, consulte nosso estudo sobre o mercado de dispositivos médicos no Brasil.
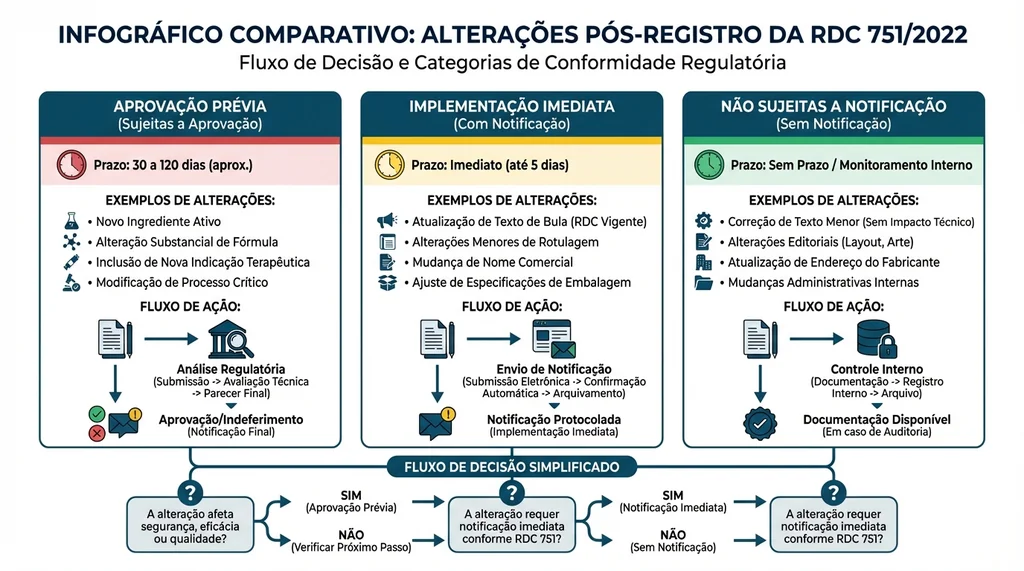
Alterações Pós-Registro: Três Categorias
A RDC 751/2022 classifica as alterações em registros e notificações em três categorias, com obrigações distintas para cada uma:
Alterações que Exigem Aprovação Prévia
Qualquer alteração que impacte segurança, eficácia ou identidade do produto requer petição formal antes da implementação. Exemplos: mudança de finalidade prevista, novo fabricante, alteração de princípio de funcionamento, novo material em contato com paciente.
Descrição completa da imagem
Infográfico das três categorias de alterações pós-registro RDC 751/2022 com exemplos e fluxos de aprovação ANVISA dispositivos médicos.
Alterações de Implementação Imediata com Notificação
Mudanças que não afetam segurança ou eficácia, mas que devem ser comunicadas à ANVISA no mesmo momento da implementação. Exemplos: nova versão de software sem impacto clínico, alteração de fornecedor de embalagem sem mudança no material.
Alterações Não Sujeitas a Notificação
Mudanças de caráter administrativo ou estético que não precisam ser comunicadas. Devem, no entanto, ser documentadas internamente no arquivo do produto. Exemplos: atualização do logotipo da empresa, mudança de endereço.
ANVISA vs FDA vs EU MDR: Comparativo Regulatório
A convergência regulatória é uma tendência global. A RDC 751/2022 posiciona o Brasil entre os sistemas mais alinhados internacionalmente:
| Aspecto | ANVISA (RDC 751/2022) | FDA (EUA) | EU MDR |
|---|---|---|---|
| Número de regras | 22 | Não aplica (510k/PMA) | 22 (mesmas) |
| Número de classes | 4 (I, II, III, IV) | 3 (I, II, III) | 4 (I, IIa, IIb, III) |
| Equivalência de classes | I = I | II = IIa | III = IIb | IV = III | I ≈ I | II ≈ II | III ≈ III | Referência |
| Via de reliance | AREE (IN 290/2024) | Não formal | Não formal |
| SaMD | Regra 11 | Digital Health Center of Excellence | Regra 11 (MDCG 2019-11) |
| UDI | SIUD (2025–2028) | UDI (implementado) | EUDAMED (em curso) |
| Boas práticas | RDC 665/2022 + ISO 13485 | 21 CFR Part 820 (QMSR) | ISO 13485 + IVDR Annex IX |
A harmonização com o EU MDR é particularmente relevante: empresas que já possuem marcação CE podem usar a documentação europeia como base para a submissão AREE no Brasil, reduzindo significativamente o tempo e custo de elaboração do dossiê. Veja também nosso guia completo sobre ANVISA e dispositivos médicos em 2026.
Descontos na TFVS e Redução de Custos
A Taxa de Fiscalização de Vigilância Sanitária (TFVS) é o principal custo direto do processo regulatório. A RDC 751/2022, combinada com a legislação tributária, prevê descontos escalonados conforme o porte da empresa:
- Empresas com receita bruta até R$ 6 milhões/ano: desconto de 60% na TFVS.
- Empresas de pequeno porte (EPP): desconto de aproximadamente 90%.
- Microempresas (ME): desconto de aproximadamente 95%.
- Startups certificadas pelo MCTIC: podem acumular benefícios com editais de inovação em saúde.
Para uma startup de base tecnológica desenvolvendo um SaMD Classe II, o custo total de notificação pode ser inferior a R$ 150, considerando os descontos disponíveis, tornando o acesso ao mercado regulado muito mais viável para empresas emergentes.
RDC 848/2024: Requisitos Essenciais de Segurança
Publicada em 2024, a RDC 848/2024 complementa a RDC 751/2022 estabelecendo os Requisitos Essenciais de Segurança e Performance (RESD), equivalentes ao ESPR europeu. O fabricante deve demonstrar que o dispositivo atende a esses requisitos antes de qualquer submissão regulatória. A conformidade é demonstrada preferencialmente por meio de normas técnicas harmonizadas (ABNT, IEC, ISO), cujo uso cria presunção de conformidade, reduzindo o ônus probatório no dossiê.
Perguntas Frequentes
Quais produtos estão dispensados de registro ou notificação pela RDC 751/2022?
O Anexo I da RDC 751/2022 lista os produtos dispensados, que correspondem basicamente aos itens que constavam na antiga RE 1.554/2002. Incluem produtos como óculos de leitura simples sem prescrição, termômetros ambientais e alguns equipamentos de uso exclusivamente veterinário. A consulta ao Anexo I deve ser o primeiro passo antes de iniciar qualquer processo regulatório.
Como funciona a via AREE e quais países são aceitos como referência?
A AREE (regulamentada pela IN 290/2024) permite que a ANVISA utilize a decisão regulatória de autoridades estrangeiras como principal evidência de análise. Os países aceitos são: EUA (FDA), Canadá (Health Canada), Austrália (TGA) e Japão (PMDA). O fabricante deve ter aprovação vigente nesses países e submeter a documentação utilizada na aprovação estrangeira, traduzida para o português. O processo reduz o tempo de análise em aproximadamente 30%.
O que é o CBPF e quando ele é obrigatório?
O Certificado de Boas Práticas de Fabricação (CBPF) é o documento que atesta que a planta produtiva do fabricante opera em conformidade com as boas práticas regulatórias. É obrigatório para dispositivos das Classes III e IV, tanto para fabricantes nacionais quanto importados. Para fabricantes estrangeiros, o certificado MDSAP é o mais aceito pela ANVISA, cobrindo simultaneamente Brasil, EUA, Canadá, Austrália e Japão.
Como classificar um software como dispositivo médico pela Regra 11?
A Regra 11 classifica SaMD com base no impacto clínico da informação gerada pelo software. Softwares que fornecem informações apenas para fins de armazenamento ou exibição são Classe I. Softwares que geram diagnóstico ou orientam tratamento em condições de risco moderado são Classe II ou III. Softwares que substituem o julgamento clínico em situações de risco de vida são Classe IV. O IMDRF/SaMD N41 e o guia MDCG 2019-11 são as referências internacionais recomendadas para a análise.
Qual é o prazo real para aprovação de um registro Classe III na ANVISA?
O prazo legal é de 250 a 365 dias corridos após a aceitação da petição. No entanto, o prazo mediano real observado é de aproximadamente 389 dias, e pode superar 500 dias em casos de exigências (solicitações de complementação de documentos). A principal estratégia para reduzir esse prazo é submeter um dossiê técnico completo e sem inconsistências na primeira submissão, o que exige domínio dos requisitos da norma e das guias de perguntas e respostas publicadas pela ANVISA.
Conclusão
A RDC 751/2022 representa uma maturidade regulatória significativa para o Brasil, alinhando o país às melhores práticas internacionais em um setor que movimenta US$ 15,28 bilhões e emprega mais de 85.000 profissionais. Para profissionais de regulatory affairs e engenheiros de qualidade, o domínio das 22 regras de classificação, da estrutura IMDRF do dossiê e das vias alternativas como a AREE é condição essencial para atuar com eficiência nesse mercado. Uma compreensão sólida de como funciona a ANVISA e seu papel na regulamentação de saúde também é fundamental para navegar o sistema regulatório brasileiro.
Para aprofundar seu conhecimento em regulamentação de dispositivos médicos, acesse nosso guia completo sobre ANVISA e dispositivos médicos e o conteúdo sobre boas práticas de fabricação (RDC 665/2022).
Fontes externas de referência: ANVISA, RDC 751/2022 (publicação oficial) Portal ANVISA, Dispositivos Médicos e IMDRF, Table of Contents for Technical Documentation.
Artigo elaborado pela equipe editorial do Portal Engenharia Biomédica, com base na RDC 751/2022, IN 290/2024, RDC 848/2024 e publicações técnicas da ANVISA e do IMDRF. Atualizado em fevereiro de 2026.
Artigos relacionados
ANVISA e Dispositivos Médicos: Guia Completo de Regulamentação [Atualizado 2026]
Guia completo sobre regulamentação de dispositivos médicos pela ANVISA: classes de risco I-IV, fluxo de registro, documentação exigida, prazos, custos e as principais RDCs de 2022 a 2025. Conteúdo atualizado para profissionais de regulatory affairs, startups e fabricantes.
Certificação INMETRO para Equipamentos Médicos: Processo Passo a Passo
Guia completo sobre a certificação INMETRO de equipamentos eletromédicos no Brasil: Portaria 384/2020, normas IEC 60601, laboratórios acreditados, custos, prazos e o processo detalhado passo a passo para fabricantes e importadores.
Como Registrar um Dispositivo Médico na ANVISA: Roteiro Prático para Startups e Fabricantes
Guia completo do processo de registro de dispositivos médicos na ANVISA: classificação, CBPF, ensaios técnicos, submissão no sistema SOLICITA e acompanhamento. Prazos reais, custos atualizados e erros mais comuns a evitar.
IEC 60601: A Norma de Segurança de Equipamentos Eletromédicos Que Todo Engenheiro Precisa Conhecer
Entenda a estrutura completa da IEC 60601, o modelo sanduíche de normas gerais, colaterais e particulares, os conceitos de MOPP/MOOP, classificação de partes aplicadas e os requisitos de ensaio que definem a segurança de equipamentos eletromédicos no Brasil e no mundo.