Registrar um dispositivo médico na ANVISA é um dos processos mais exigentes que uma startup ou fabricante pode enfrentar no mercado brasileiro. O caminho correto depende fundamentalmente da classe de risco do produto: enquanto dispositivos de baixo risco (Classe I e II) podem ser notificados em cerca de 30 dias, equipamentos de alta complexidade (Classe III e IV) exigem certificação de Boas Práticas de Fabricação (CBPF), ensaios técnicos rigorosos e podem levar até quatro anos para obter autorização de comercialização. Conhecer cada etapa antes de começar evita retrabalho custoso e atrasos que comprometem o plano de negócios.
Este artigo integra nosso Guia Completo de Dispositivos Médicos na ANVISA 2026 e detalha o workflow operacional do registro, da classificação à pós-comercialização. Para entender a base legal que estrutura todo este processo, consulte também nossa análise da RDC 751/2022.
Visão Geral do Processo: Notificação ou Registro?
A primeira decisão estratégica é identificar qual via regulatória se aplica ao seu produto. A RDC 751/2022 estabelece quatro classes de risco, e a via de autorização varia conforme a classe:
| Classe | Risco | Via | Prazo estimado | CBPF obrigatório? |
|---|---|---|---|---|
| I | Mínimo | Notificação | ~30 dias | Não |
| II | Baixo | Notificação | ~30 dias | Não |
| III | Médio-alto | Registro | 12–24 meses | Sim |
| IV | Alto | Registro | 18–48 meses | Sim |
A notificação funciona como um ato declaratório: a empresa apresenta a documentação e, salvo irregularidades identificadas no prazo de análise, o produto fica automaticamente autorizado. O registro, por outro lado, envolve análise ativa pela ANVISA, que pode formular exigências técnicas durante o processo. Para compreender como a agência se organiza internamente para conduzir essas análises, veja nosso artigo sobre a estrutura e as atribuições da ANVISA.
A ANVISA reconheceu oficialmente em 2025 um aumento significativo das filas de análise, atribuído à redução de 30% do quadro de servidores nos últimos 10 anos, ao aumento na complexidade tecnológica dos produtos e ao crescimento no volume de pedidos.
— ANVISA, Edital de Chamamento 22/2025, 2025
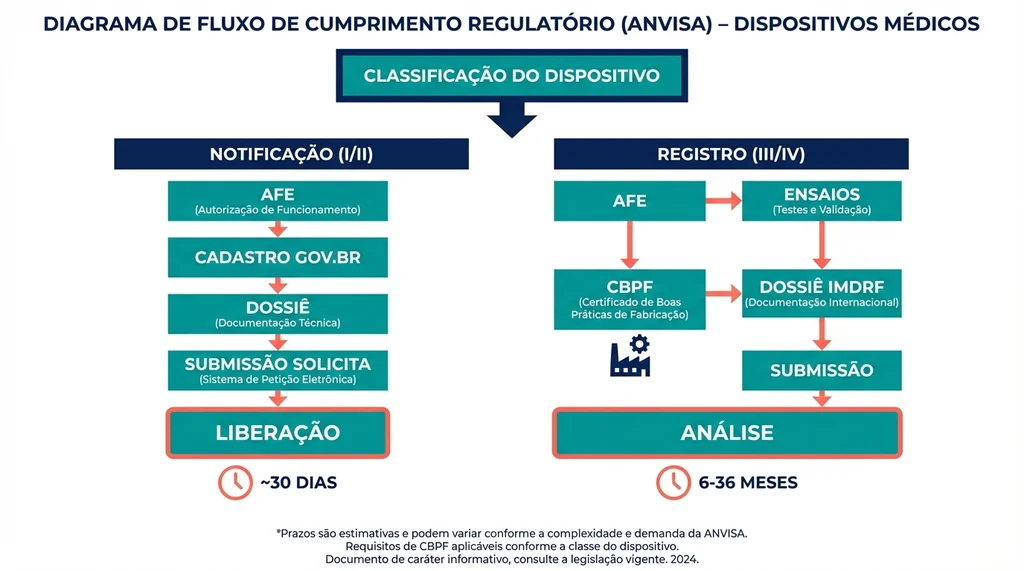
Descrição completa da imagem
Fluxograma com dois caminhos de registro na ANVISA: Notificação para dispositivos Classe I e II e Registro para Classe III e IV, com etapas, prazos e requisitos de CBPF para cada via regulatória..
Pré-Requisitos Antes de Qualquer Submissão
Independentemente da classe do dispositivo, quatro habilitações precisam estar em ordem antes de protocolar qualquer pedido no sistema SOLICITA:
1. Autorização de Funcionamento de Empresa (AFE)
A AFE é concedida pela ANVISA e habilita a empresa a fabricar, importar, exportar ou distribuir produtos regulados. Para obtê-la, é necessário cadastro no sistema Gov.br, pagamento da TFVS de AFE e designação de Responsável Técnico (RT) com formação compatível com os produtos da empresa. A AFE deve estar ativa e válida durante todo o processo de registro.
2. Cadastro no Gov.br e no Sistema SOLICITA
O sistema SOLICITA é a plataforma eletrônica da ANVISA para submissão e acompanhamento de petições. O acesso requer conta Gov.br com nível de autenticação "Prata" ou "Ouro". Orientamos que o Responsável Técnico e os representantes legais realizem esse cadastro com antecedência, pois a validação pode levar dias úteis.
3. Certificado de Boas Práticas de Fabricação (CBPF)
Obrigatório para Classe III e IV. O CBPF atesta que o fabricante, nacional ou estrangeiro, cumpre os requisitos da RDC 665/2022 de Boas Práticas de Fabricação. Para fabricantes nacionais, a inspeção é conduzida pela própria ANVISA. Para fabricantes estrangeiros, o CBPF pode ser obtido por inspeção direta da ANVISA (prazo de cerca de 2 anos) ou por reconhecimento de certificação MDSAP (Medical Device Single Audit Program), o que reduz o prazo para cerca de 4 anos em média, conforme a RDC 850/2024.
Em 2019, 48,7% das certificações internacionais de Boas Práticas de Fabricação da ANVISA já se baseavam no MDSAP, demonstrando a consolidação do programa como via preferencial para fabricantes estrangeiros.
— ANVISA/IMDRF, Relatório MDSAP, 2019
4. Responsável Técnico (RT)
O RT deve ser um profissional habilitado pelo conselho de classe correspondente à natureza do dispositivo (engenheiro, médico, farmacêutico, fisioterapeuta, entre outros). Ele assina tecnicamente todas as petições e é pessoalmente responsável pelas informações declaradas.
Etapa 1: Classificação do Dispositivo
A classificação correta é o alicerce de todo o processo. Erros nessa etapa costumam ser descobertos apenas na análise pela ANVISA, gerando arquivamento ou exigências que reiniciam prazos. A RDC 751/2022 define regras de classificação baseadas em critérios como:
- Duração do contato com o paciente (transitório, curta duração, longa duração)
- Invasividade (não invasivo, invasivo, implantável)
- Dependência de fonte de energia (ativo ou não ativo)
- Finalidade de uso (diagnóstico, terapêutico, monitoramento)
- Contato com sistema nervoso central, circulação central ou coração
Softwares como Aplicativos de Saúde seguem regras específicas de classificação definidas para SaMD (Software as a Medical Device) que consideram a gravidade das decisões clínicas apoiadas pelo software.
Dica prática: consulte as listas de produtos exemplificados na RDC 751/2022 e, em caso de dúvida, utilize o serviço de Consulta Prévia da ANVISA (petição específica no SOLICITA), que fornece orientação formal sobre classificação e via regulatória aplicável.
Etapa 2: Ensaios Técnicos e Documentação Pré-Submissão
Para dispositivos de Classe III e IV, a preparação do dossiê técnico é a fase mais demorada e custosa. Os ensaios devem ser realizados em laboratórios acreditados pelo INMETRO ou equivalentes internacionais reconhecidos.
Ensaios de segurança elétrica
Equipamentos eletromédicos precisam atender à norma IEC 60601-1 (segurança básica e desempenho essencial) e às normas colaterais aplicáveis (IEC 60601-1-2 para compatibilidade eletromagnética, IEC 60601-1-6 para usabilidade, entre outras). Os testes incluem resistência de isolação, correntes de fuga, resistência ao fogo e EMC.
Biocompatibilidade
Dispositivos com contato com o corpo humano, mesmo indireto, precisam de avaliação de biocompatibilidade conforme a série ISO 10993. O escopo dos testes depende da natureza e duração do contato: citotoxicidade, sensibilização, irritação, pirogenicidade e genotoxicidade são os mais comuns.
Gerenciamento de riscos
O dossiê deve conter o arquivo de gerenciamento de riscos completo conforme ISO 14971:2019, incluindo plano de gerenciamento de riscos, análise de riscos, avaliação de riscos, controles implementados e relatório de gerenciamento de riscos residuais.
Evidências clínicas
A RDC 848/2024 regulamenta os requisitos de evidência clínica para dispositivos médicos no Brasil. Para Classe III e IV, geralmente é exigida avaliação clínica estruturada, que pode ser baseada em literatura científica existente, dados clínicos pré-clínicos ou estudos clínicos prospectivos quando não houver dados suficientes disponíveis na literatura.
O Dossiê Técnico: Estrutura IMDRF e Evidências Clínicas
A ANVISA adota a estrutura de Sumário Técnico de Submissão (STS) definida pelo International Medical Device Regulators Forum (IMDRF). O dossiê é organizado em seções padronizadas que facilitam a análise regulatória:
| Seção IMDRF | Conteúdo principal | Normas de referência |
|---|---|---|
| 1 – Sumário do Dispositivo | Descrição, finalidade de uso, população-alvo | RDC 751/2022 |
| 2 – Informações de Segurança e Desempenho | Dados pré-clínicos, clínicos, benef./risco | RDC 848/2024, ISO 14971 |
| 3 – Fabricação e Controle de Qualidade | Processo produtivo, controles, embalagem | RDC 665/2022 |
| 4 – Estudos de Segurança | Biocompatibilidade, esterilização, estabilidade | ISO 10993, ISO 11135 |
| 5 – Estudos de Desempenho Elétrico | Segurança elétrica, EMC, usabilidade | IEC 60601-1, IEC 62366 |
| 6 – Rotulagem | Rótulo, IFU, embalagem | RDC 751/2022, Annex IV |
Toda a documentação deve ser apresentada em português. Documentos originalmente em inglês precisam de tradução juramentada, salvo exceções previstas em norma específica. O dossiê é submetido integralmente em formato eletrônico pelo SOLICITA, com cada seção em arquivo PDF separado e tamanho máximo definido pela plataforma.

Etapa 3: Submissão no Sistema SOLICITA
O sistema SOLICITA é o portal único de peticionamento eletrônico da ANVISA. A submissão de um pedido de registro envolve as seguintes ações:
- Seleção do tipo de petição: Notificação de DM, Registro de DM, Alteração Pós-Registro ou CBPF, conforme aplicável
- Preenchimento do formulário eletrônico: dados do produto, classificação, fabricante, RT, finalidade de uso e declarações regulatórias
- Upload dos documentos: dossiê técnico completo em PDFs organizados por seção IMDRF
- Emissão do GRU: guia de recolhimento da TFVS, que deve ser paga antes da efetivação da petição
- Protocolo da petição: após o pagamento, o sistema registra o número de protocolo e a petição entra na fila de análise
Atenção: a petição somente é considerada protocolada após a confirmação do pagamento da TFVS. Petições com documentação incompleta podem ser arquivadas sumariamente, sendo necessário novo protocolo com pagamento de nova taxa.
Via AREE: Aprovação com Base em Autoridade Regulatória Estrangeira Equivalente
A Instrução Normativa 290/2024 regulamenta a via AREE, que permite à ANVISA basear-se em decisões de autoridades reconhecidas como FDA (EUA), Health Canada, TGA (Austrália) e PMDA (Japão) para aprovação de dispositivos Classe III e IV. Empresas que já possuem aprovação nessas jurisdições podem protocolar via AREE e obter redução de prazo de análise de aproximadamente 30% em relação à via convencional. A documentação exigida inclui cópia da autorização estrangeira, relatório de análise da autoridade de referência e declaração de equivalência regulatória.
Em 2024, a ANVISA recebeu aproximadamente 380 pedidos via reliance regulatório (IN 290/2024) para dispositivos médicos, indicando adoção crescente dessa via acelerada por fabricantes com aprovações em mercados internacionais.
— ANVISA, Balanço Regulatório de Dispositivos Médicos, 2024
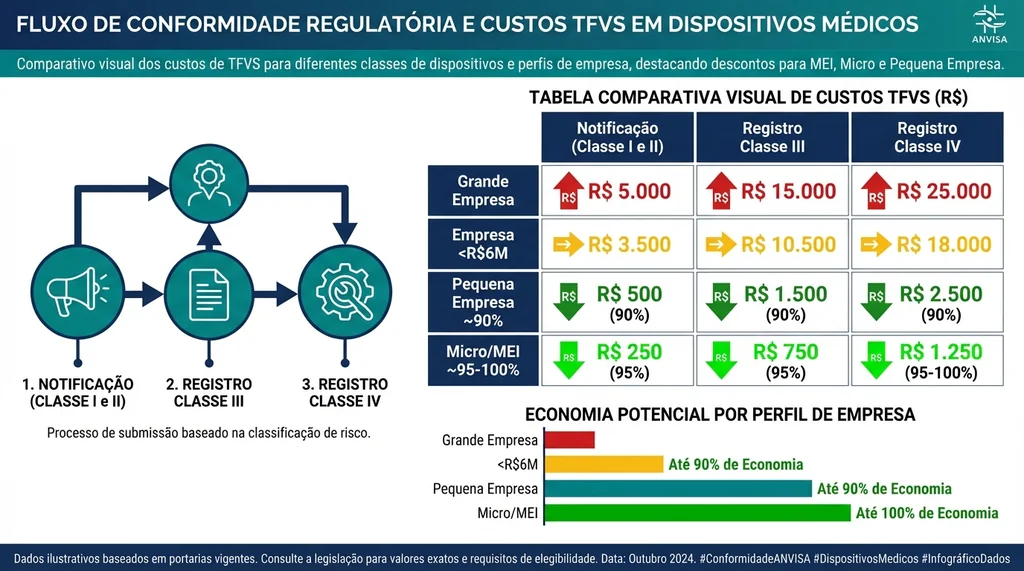
Custos e Descontos: TFVS Atualizada para 2026
A Taxa de Fiscalização de Vigilância Sanitária (TFVS) é o principal custo regulatório direto, mas está longe de representar o custo total do processo. Os valores de referência para 2026 são:
| Petição | Valor integral | Desconto <R$6M | Desconto ~90% | Desconto ~95% |
|---|---|---|---|---|
| Notificação (I/II) | R$ 1.406 | R$ 562 | R$ 141 | R$ 70 |
| Registro Classe III | R$ 8.510 | R$ 3.404 | R$ 851 | R$ 426 |
| Registro Classe IV | R$ 19.856 | R$ 7.942 | R$ 1.986 | R$ 993 |
| CBPF Estrangeiro | R$ 72.805 | R$ 29.122 | R$ 7.281 | R$ 3.640 |
Os descontos são aplicados automaticamente com base no porte da empresa declarado na Receita Federal. MEIs são isentos de TFVS, mas as obrigações regulatórias permanecem integrais. Empresas com faturamento bruto abaixo de R$ 6 milhões obtêm 60% de desconto; pequenas empresas (EPP), cerca de 90%; microempresas (ME), cerca de 95%.
Além da TFVS, o custo total de um processo de registro de Classe IV pode incluir:
- Ensaios laboratoriais: R$ 50.000 – R$ 300.000
- Consultoria regulatória especializada: R$ 30.000 – R$ 150.000
- Tradução juramentada de documentos: R$ 5.000 – R$ 30.000
- CBPF do fabricante estrangeiro (inspeção ANVISA): R$ 72.805 (taxa) + despesas de missão
- Estudos clínicos (quando necessários): variável, podendo ultrapassar R$ 1 milhão
O custo total realístico para Classe IV com fabricante estrangeiro sem certificação MDSAP situa-se entre R$ 300.000 e R$ 500.000 ao longo de todo o processo.
Prazos Realistas por Classe
Os prazos legais de análise pela ANVISA são contados em dias úteis e podem ser suspensos durante períodos de exigência. O que os fabricantes experienciam na prática é frequentemente superior aos prazos nominais:
| Classe | Prazo legal (dias úteis) | Prazo real (meses) | Principal gargalo |
|---|---|---|---|
| I / II (Notificação) | 30 | 1–6 | Documentação incompleta |
| III (Registro) | 365 | 12–24 | Exigências técnicas, CBPF |
| IV (Registro) | 730 | 18–48 | Ensaios clínicos, CBPF estrangeiro |
| Via AREE (III/IV) | 365 | 10–30 | Equivalência regulatória |
O prazo de análise pode ser suspenso por até 60 dias úteis em cada exigência técnica formulada pela ANVISA. Empresas que respondem às exigências de forma completa e ágil na primeira oportunidade reduzem significativamente o prazo total.
Acompanhamento, Exigências e Pós-Registro
Monitoramento no SOLICITA
Após o protocolo, todas as movimentações da petição são disponibilizadas no sistema SOLICITA. É fundamental verificar periodicamente o status, pois exigências técnicas têm prazo de resposta definido e o não atendimento implica o arquivamento da petição sem restituição da taxa.
Resposta a Exigências
Exigências técnicas são notificações formais em que a ANVISA solicita documentos adicionais, esclarecimentos ou correções no dossiê. A resposta deve ser protocolada no prazo estipulado (geralmente 60 dias úteis), diretamente no SOLICITA, com todos os documentos solicitados devidamente assinados pelo RT.
Alterações Pós-Registro
Após a concessão do registro (validade de 10 anos), qualquer modificação no produto, processo ou rotulagem deve ser comunicada à ANVISA conforme a tipologia de alteração:
- Com petição de aprovação prévia: mudanças que afetam segurança, desempenho ou finalidade de uso
- Com petição de notificação imediata: mudanças administrativas de menor impacto
- Não reportáveis: melhorias sem impacto em segurança ou desempenho, documentadas internamente
Revalidação do Registro
O registro tem validade de 10 anos e deve ser revalidado no primeiro semestre do último ano de vigência. A não revalidação no prazo implica o cancelamento automático do registro e a necessidade de novo processo completo.
Erros Mais Comuns e Como Evitá-los
Com base em processos analisados e na experiência de profissionais de assuntos regulatórios, estes são os erros que mais frequentemente atrasam ou inviabilizam registros na ANVISA:
1. Classificação incorreta do dispositivo
Classificar o produto em classe inferior à correta é o erro mais grave: além do arquivamento da petição, pode configurar infração sanitária. Use a Consulta Prévia da ANVISA sempre que houver dúvida fundada.
2. CBPF vencido ou com escopo inadequado
O CBPF deve ter escopo que cubra exatamente o produto sendo registrado. Um CBPF válido para "equipamentos de monitorização" não cobre automaticamente "bombas de infusão implantáveis". Verifique o escopo antes de submeter.
3. Documentação sem assinatura do Responsável Técnico
Todos os documentos técnicos, declarações e formulários da petição precisam de assinatura eletrônica qualificada do RT. Documentos sem assinatura válida geram exigência imediata.
4. Laudos de ensaios fora da validade ou emitidos por laboratório sem acreditação
A ANVISA exige que os ensaios sejam realizados por laboratórios acreditados pelo INMETRO (RBLE/RNLA) ou equivalentes. Laudos com mais de 5 anos podem não ser aceitos dependendo do tipo de ensaio.
5. Tradução juramentada ausente ou com tradutor sem habilitação
Documentos em idioma estrangeiro precisam de tradução juramentada por tradutor público juramentado registrado na Junta Comercial do estado. Traduções não juramentadas, mesmo de alta qualidade técnica, não são aceitas.
Perguntas Frequentes sobre Registro na ANVISA
Startups em fase pré-receita podem se qualificar para descontos na TFVS?
Sim. O desconto de TFVS é calculado com base no faturamento bruto declarado à Receita Federal no último exercício. Uma startup sem faturamento ou com faturamento abaixo de R$ 6 milhões se qualifica automaticamente para 60% de desconto. MEIs são isentos da TFVS. O importante é que a empresa esteja regularmente constituída e o CNPJ ativo.
É possível comercializar o dispositivo enquanto o registro está em análise?
Não, para Classe III e IV. Para Classe I e II na via de Notificação, a comercialização pode ocorrer após o protocolo, mas antes da análise, o que representa um risco regulatório caso irregularidades sejam identificadas posteriormente. Em caráter excepcional, a ANVISA pode conceder autorização especial temporária em situações de emergência em saúde pública.
Qual é a diferença prática entre MDSAP e a inspeção direta da ANVISA para obter o CBPF?
A inspeção direta da ANVISA para CBPF de fabricante estrangeiro leva em média 2 anos, considerando o agendamento, a própria inspeção e a emissão do certificado. O MDSAP é um programa de auditoria única que é reconhecido por Brasil, EUA, Canadá, Austrália e Japão simultaneamente. Para o CBPF via MDSAP, a RDC 850/2024 estabelece prazo de análise de 120 dias úteis após a submissão dos relatórios do MDSAP. Para fabricantes que já possuem ou planejam obter aprovação em múltiplas jurisdições, o MDSAP representa uma economia significativa de tempo e custo.
O que acontece se minha petição for arquivada?
O arquivamento ocorre por não atendimento de exigência no prazo, pagamento de TFVS insuficiente ou documentação fundamentalmente inadequada. A TFVS não é restituída. O fabricante pode protocoliar nova petição a qualquer momento, com pagamento de nova taxa. Em alguns casos, os documentos previamente submetidos podem ser reaproveitados, desde que ainda estejam dentro do prazo de validade.
Existe um caminho mais rápido para dispositivos já aprovados nos EUA ou Europa?
Sim. A via AREE (IN 290/2024) permite que a ANVISA utilize como referência a análise técnica de autoridades como FDA, Health Canada, TGA e PMDA. Para Classe III e IV com aprovação nessas jurisdições, a AREE pode reduzir o prazo de análise em até 30%. A documentação exigida inclui a autorização estrangeira vigente, o relatório de avaliação da autoridade de referência e o dossiê técnico na estrutura IMDRF. Certificação CE europeia isoladamente não é aceita como referência na via AREE, mas pode compor evidências no dossiê técnico convencional. Confira também nosso estudo de caso sobre como empresas internacionais obtêm aprovação FDA, uma etapa que pode facilitar o registro brasileiro via AREE.
Para aprofundar seu entendimento sobre o ecossistema regulatório de dispositivos médicos no Brasil, acesse nosso guia completo atualizado para 2026 e acompanhe as publicações da ANVISA para estar sempre informado sobre mudanças regulatórias que impactem seu produto. Para dúvidas específicas sobre seu dispositivo, a seção de perguntas frequentes da ANVISA e a modalidade de Consulta Prévia são os canais oficiais recomendados. A biblioteca de documentos do IMDRF disponibiliza gratuitamente todos os guias de estrutura de dossiê técnico adotados como referência no Brasil.
Artigos relacionados
ANVISA e Dispositivos Médicos: Guia Completo de Regulamentação [Atualizado 2026]
Guia completo sobre regulamentação de dispositivos médicos pela ANVISA: classes de risco I-IV, fluxo de registro, documentação exigida, prazos, custos e as principais RDCs de 2022 a 2025. Conteúdo atualizado para profissionais de regulatory affairs, startups e fabricantes.
Certificação INMETRO para Equipamentos Médicos: Processo Passo a Passo
Guia completo sobre a certificação INMETRO de equipamentos eletromédicos no Brasil: Portaria 384/2020, normas IEC 60601, laboratórios acreditados, custos, prazos e o processo detalhado passo a passo para fabricantes e importadores.
IEC 60601: A Norma de Segurança de Equipamentos Eletromédicos Que Todo Engenheiro Precisa Conhecer
Entenda a estrutura completa da IEC 60601, o modelo sanduíche de normas gerais, colaterais e particulares, os conceitos de MOPP/MOOP, classificação de partes aplicadas e os requisitos de ensaio que definem a segurança de equipamentos eletromédicos no Brasil e no mundo.
ISO 13485 para Dispositivos Médicos: Implementação Prática do Sistema de Gestão da Qualidade
Guia completo de implementação da ISO 13485:2016 com checklists por cláusula, templates de procedimentos, custos reais de certificação e estratégias para auditoria. Essencial para gestores de qualidade e consultores do setor de dispositivos médicos no Brasil.