Software como Dispositivo Médico (SaMD) é hoje um dos segmentos de maior crescimento em toda a cadeia de tecnologia em saúde. Aplicativos de diagnóstico por imagem, algoritmos de triagem clínica, sistemas de apoio à decisão baseados em inteligência artificial, todos esses produtos precisam passar por um processo regulatório específico antes de chegar ao mercado. No Brasil, a ANVISA publicou a RDC 657/2022, estabelecendo o marco regulatório nacional e alinhando o país ao framework internacional do IMDRF.
Este artigo faz parte do Guia Definitivo de Engenharia Biomédica.
O Que é Software como Dispositivo Médico (SaMD)?
Descrição completa da imagem
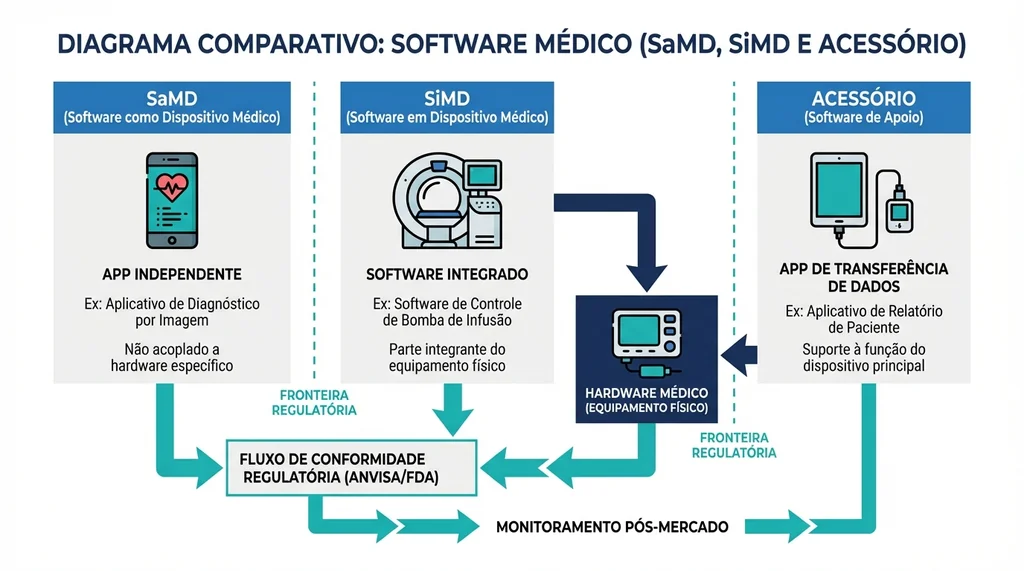
Diagrama comparativo entre SaMD, SiMD e software acessório de dispositivo médico com exemplos e fronteiras regulatórias definidas pela ANVISA e IMDRF.
A definição oficial adotada pela ANVISA, alinhada ao IMDRF (International Medical Device Regulators Forum), estabelece que SaMD é "software destinado a ser usado para uma ou mais finalidades médicas sem fazer parte de um dispositivo médico de hardware". Em outras palavras, o software funciona de forma independente, não precisa de um equipamento físico específico para operar e, por si só, cumpre uma função diagnóstica, terapêutica, de monitoramento ou de previsão clínica.
Essa definição exclui dois tipos de software frequentemente confundidos com SaMD:
- Software in Medical Device (SiMD): software que faz parte integrante de um dispositivo médico físico, como o firmware de um ventilador mecânico ou de um monitor de ECG. Esse tipo é regulado como parte do próprio equipamento.
- Software acessório de dispositivo médico: software que auxilia na operação de um dispositivo já regulado, mas que não executa função médica autônoma, por exemplo, um aplicativo de transferência de dados de um glucômetro para o celular, sem interpretar os resultados.
A fronteira entre essas categorias é o ponto mais crítico para o profissional de engenharia biomédica: um mesmo aplicativo pode ser SaMD, SiMD ou acessório dependendo da finalidade declarada e da relação com o hardware.
RDC 657/2022: O Marco Regulatório Brasileiro
Publicada em dezembro de 2022, a RDC 657 foi o passo decisivo da ANVISA para criar um regime regulatório específico para SaMD. Antes dessa resolução, softwares médicos eram enquadrados nas regras gerais de dispositivos médicos, sem considerar as particularidades do desenvolvimento ágil, das atualizações contínuas e dos modelos de machine learning.
Os principais pontos da RDC 657/2022 incluem:
- Adoção formal da definição IMDRF de SaMD
- Critérios para distinguir SaMD de software de bem-estar (wellness) não regulado
- Base para a matriz de risco e classificação por classes regulatórias
- Requisitos de ciclo de vida de software baseados nas normas IEC 62304 e IEC 82304
- Obrigações de pós-mercado, incluindo vigilância e atualização de versões
A resolução está atualmente em processo de revisão pela ANVISA, com foco em dois temas emergentes: o tratamento de software adaptável (aquele que aprende e muda com o tempo, como modelos de IA) e a incorporação do conceito de PCCP, Predetermined Change Control Plan já adotado pelo FDA americano para gerenciar atualizações de algoritmos sem exigir uma nova submissão regulatória a cada mudança.
Para aprofundar o entendimento sobre o ecossistema regulatório brasileiro, consulte nosso artigo sobre regulamentação ANVISA para dispositivos médicos.
A Matriz de Risco IMDRF: Como Classificar um SaMD
Descrição completa da imagem
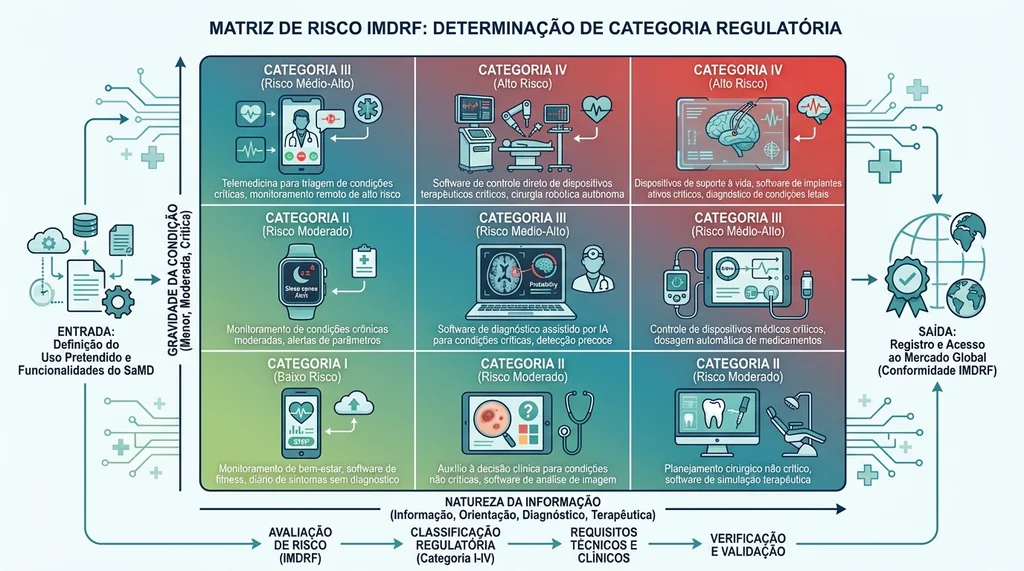
Matriz de risco IMDRF 3x3 mostrando categorias I a IV para classificação de SaMD com base em natureza da informação gerada e gravidade da condição clínica.
O framework IMDRF propõe uma matriz 3×3 para determinar o nível de risco de um SaMD. A combinação entre a natureza da informação produzida e a gravidade da condição do paciente resulta em quatro categorias (I a IV), onde IV representa o maior risco.
| Natureza da Informação / Gravidade da Condição | Não Grave | Grave | Crítica / Fatal |
|---|---|---|---|
| Informar (ex.: registrar dados) | Cat. I | Cat. I | Cat. II |
| Direcionar (ex.: apoiar diagnóstico) | Cat. I | Cat. II | Cat. III |
| Diagnosticar/Tratar (ex.: fechar diagnóstico automaticamente) | Cat. II | Cat. III | Cat. IV |
Um algoritmo que detecta automaticamente hemorragia craniana em tomografias e aciona o fluxo de emergência sem intervenção humana intermediária, por exemplo, estaria na Categoria IV: diagnóstico autônomo em condição crítica. Já um aplicativo que registra sintomas de uma condição não grave e sugere ao médico uma hipótese diagnóstica seria Categoria I ou II.
Classificação ANVISA: Regra 11 e Classes I–IV
No Brasil, a classificação de SaMD é feita pela Regra 11 da RDC 751/2022 que transpõe a lógica IMDRF para o sistema nacional de classes de risco (I a IV). As implicações práticas de cada classe são significativas:
| Classe | Via Regulatória | Requisitos Principais | Prazo Estimado |
|---|---|---|---|
| Classe I | Notificação simplificada | Cadastro básico, declaração de conformidade | 30–60 dias |
| Classe II | Notificação simplificada | Documentação técnica, rotulagem, IEC 62304 | 60–120 dias |
| Classe III | Registro completo | Dossiê técnico completo, estudos clínicos, CBPF | 12–24 meses |
| Classe IV | Registro completo | Dossiê técnico completo, evidências clínicas robustas, CBPF, auditorias | 24–36 meses |
O CBPF (Certificado de Boas Práticas de Fabricação) exigido para as classes III e IV é um dos principais gargalos para startups de saúde digital no Brasil. Ele requer que o fabricante, ou o desenvolvedor de software, no caso de SaMD, demonstre conformidade com processos de qualidade auditáveis, equivalentes ao que a ISO 13485 estabelece para fabricantes de dispositivos físicos.
A RDC 657/2022 foi resultado de processo que incluiu Consulta Pública n. 1.035/2021, com mais de 440 contribuições da sociedade, e Analise de Impacto Regulatorio que reconheceu a impossibilidade de aplicar as regulacoes vigentes de hardware a software.
— ANVISA, Consulta Publica 1.035/2021 e AIR da RDC 657/2022
Cenário Atual: Brasil vs. FDA vs. União Europeia
Descrição completa da imagem
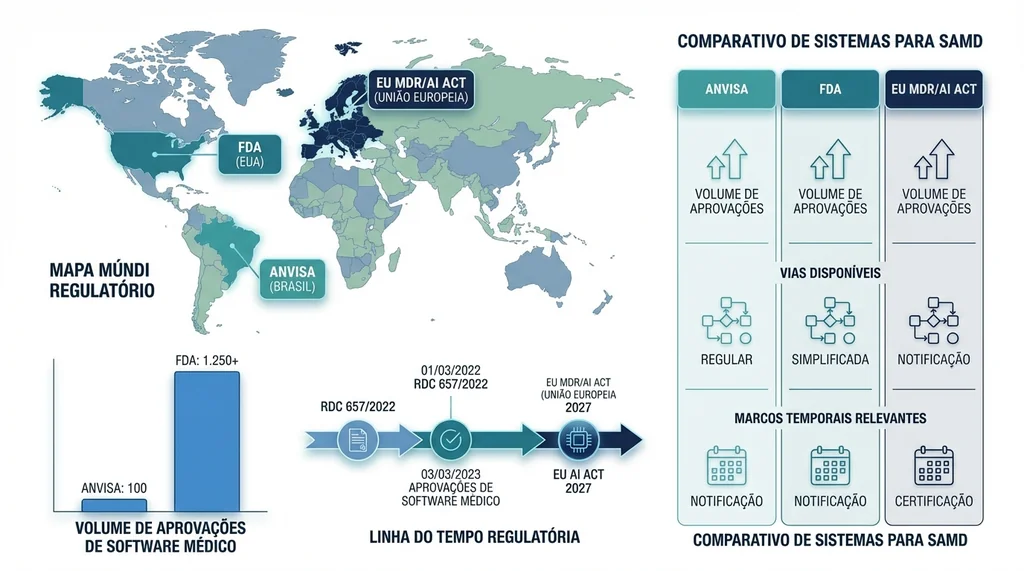
Comparação entre os sistemas regulatórios ANVISA, FDA e EU MDR para aprovação de SaMD mostrando volume de aprovações e principais marcos regulatórios de cada jurisdição.
O Brasil ainda apresenta um gap considerável em relação às autoridades regulatórias mais maduras. Enquanto a ANVISA soma aproximadamente 100 softwares e soluções de IA aprovados o FDA americano já autorizou mais de 1.250 dispositivos de IA/ML um número que cresce a uma taxa superior a 150 novos produtos por ano.
A FDA lidera mundialmente em regulacao de SaMD com IA, tendo autorizado mais de 1.250 dispositivos medicos com IA/ML ate meados de 2025, dos quais 295 somente em 2025. A maioria absoluta (97%) e aprovada via 510(k).
— FDA Digital Health Center of Excellence, 2025
Cada jurisdição tem sua via regulatória específica:
- FDA (EUA): utiliza três vias principais, 510(k) para dispositivos com predicate device equivalente, De Novo para tecnologias sem precedente, e PMA (Premarket Approval) para dispositivos de alto risco. O FDA também lidera globalmente na regulação de IA/ML com o framework AI Action Plan.
- União Europeia: o MDR (Medical Device Regulation) regula SaMD como dispositivos médicos convencionais. A partir de agosto de 2027, entra em vigor a dupla conformidade obrigatória com o EU AI Act que impõe requisitos adicionais de transparência, explicabilidade e supervisão humana para sistemas de IA classificados como alto risco.
- ANVISA: marco regulatório mais recente (RDC 657/2022), com processo de revisão em andamento e sandbox regulatório ainda em fase incipiente.
Para desenvolvedores com ambições de mercado global, a estratégia mais eficiente costuma ser iniciar pela aprovação FDA ou CE Mark e depois adaptar o dossiê técnico para a ANVISA, aproveitando o reconhecimento mútuo parcial que o Brasil vem desenvolvendo com essas jurisdições. Para entender os requisitos mais recentes do FDA, incluindo o novo QMSR e as exigências de segurança digital, veja nosso artigo sobre QMSR e cibersegurança da FDA para dispositivos médicos.
Exemplos Práticos de SaMD no Mercado
Alguns casos reais ilustram bem o espectro de aplicações e o nível de risco regulatório envolvido:
- Aidoc (hemorragia craniana): algoritmo de IA que analisa tomografias computadorizadas e detecta automaticamente sinais de hemorragia intracraniana, priorizando a fila de leitura radiológica. Aprovado pelo FDA como Classe II via 510(k). No contexto IMDRF, seria Categoria III ou IV por diagnosticar condição crítica.
- Laura (deterioração clínica): sistema de monitoramento contínuo desenvolvido no Brasil que analisa sinais vitais de pacientes hospitalizados e alerta para risco de deterioração clínica. Exemplo de SaMD nacional que direcionou condição grave, Classe III pela Regra 11.
- brain4care: empresa brasileira com tecnologia SaMD para monitoramento não invasivo de pressão intracraniana. Caso de sucesso de desenvolvimento regulatório nacional com expansão internacional.
Esses exemplos mostram que o segmento de SaMD não se limita a grandes corporações, startups brasileiras já estão competindo globalmente, desde que naveguem corretamente o labirinto regulatório.
LGPD e Proteção de Dados em SaMD
Qualquer SaMD que processe dados de saúde está automaticamente sujeito à Lei Geral de Proteção de Dados (LGPD). Dados de saúde são classificados como dados pessoais sensíveis exigindo base legal específica para tratamento, em geral, o consentimento do titular ou o interesse legítimo em contexto de saúde pública.
O Art. 20 da LGPD estabelece o direito do titular de solicitar revisão de decisões tomadas unicamente com base em tratamento automatizado. Para SaMDs que produzem saídas diagnósticas ou terapêuticas sem intervenção humana, isso representa um requisito de design: o sistema precisa ser construído para viabilizar essa revisão, o que reforça a necessidade de explicabilidade dos algoritmos.
Além disso, o PL 2338/2023, Marco Legal da IA classifica sistemas de IA aplicados à saúde como de alto risco impondo obrigações adicionais de transparência, registro de logs, avaliação de impacto e supervisão humana. O alinhamento com esse projeto de lei, que caminha para aprovação, deve ser parte do planejamento regulatório de qualquer novo SaMD.
Normas Técnicas Essenciais para Desenvolvimento de SaMD
O desenvolvimento de um SaMD aprovável exige conformidade com um conjunto de normas técnicas internacionais. As principais são:
| Norma | Escopo | Aplicação |
|---|---|---|
| IEC 62304 | Ciclo de vida de software médico | Obrigatória para SaMD Classes II–IV |
| IEC 82304-1 | Segurança de produtos de software de saúde | Complementar à IEC 62304, foco em app independente |
| ISO 14971 | Gerenciamento de riscos | Exigida em todas as classes, inclui riscos de IA |
| ISO 13485 | Sistema de gestão da qualidade | Base para o CBPF exigido nas classes III–IV |
| IEC 81001-5-1 | Cibersegurança em software de saúde | Crescente exigência em mercados globais |
Para um panorama completo das normas aplicáveis ao setor, acesse nosso artigo sobre normas técnicas em engenharia biomédica.
DTx: Terapêuticas Digitais, A Próxima Fronteira
As Terapêuticas Digitais (DTx) representam uma subclasse de SaMD ainda mais desafiadora do ponto de vista regulatório: são softwares que entregam intervenções terapêuticas baseadas em evidências diretamente ao paciente, sem necessariamente envolver um profissional de saúde como intermediário. Exemplos incluem aplicativos para tratamento de insônia (como o Somryst, aprovado pelo FDA), aplicativos de reabilitação cognitiva e programas digitais para dependência química.
No Brasil, as DTx ainda carecem de um caminho regulatório claro. A RDC 657/2022 cobre o aspecto de segurança e eficácia como SaMD, mas questões como reembolso pelo SUS, prescrição digital e integração com planos de saúde ainda não têm regulamentação consolidada. É um espaço de alta oportunidade e, simultaneamente, de alta incerteza regulatória.
Veja como as tendências emergentes, incluindo DTx, estão moldando o futuro da área em nosso artigo sobre tendências e futuro da engenharia biomédica.
Mercado de Saúde Digital no Brasil: Oportunidade e Desafio
O mercado de saúde digital no Brasil foi avaliado em US\$ 6,35 bilhões e cresce a uma taxa anual composta (CAGR) de aproximadamente 23%, impulsionado pela pandemia de COVID-19, pelo avanço da telemedicina regulada e pelo crescente investimento em startups de healthtech. Esse crescimento cria uma pressão direta sobre a capacidade regulatória da ANVISA e sobre a formação de engenheiros biomédicos com expertise em regulação de software.
O ecossistema brasileiro conta com mais de 1.200 healthtechs ativas, crescimento de 35% desde 2020, das quais 130 aplicam inteligencia artificial. O mercado de saude digital pode alcancar USD 21,9 bilhoes ate 2030.
— Distrito Healthtech Report 2024; Grand View Research, 2024
O sandbox regulatório, mecanismo que permite testar produtos inovadores em ambiente controlado antes do registro formal, ainda está em fase incipiente na ANVISA, ao contrário do que ocorre na MHRA britânica e na própria FDA. A expectativa do setor é que a revisão da RDC 657 inclua um programa estruturado de sandbox para SaMD, reduzindo o tempo de aprovação para produtos de baixo risco e estimulando a inovação nacional.
Perguntas Frequentes sobre SaMD
Um aplicativo de calculadora de dose de medicamento é SaMD?
Depende da finalidade declarada. Se o aplicativo apenas exibe uma fórmula de cálculo sem interpretar o resultado ou recomendar conduta clínica, pode ser considerado uma ferramenta de referência não regulada. Se ele calcula a dose individualizada com base em parâmetros do paciente e recomenda a dose específica ao prescritor, passa a ser SaMD, provavelmente Classe II ou III, dependendo da gravidade da medicação envolvida.
A ANVISA exige estudos clínicos para aprovar SaMD?
Para classes I e II, evidências clínicas podem ser baseadas em literatura científica existente e dados de desempenho do software. Para classes III e IV, a ANVISA exige estudos clínicos com metodologia robusta, podendo ser estudos retrospectivos em bases de dados reais ou prospectivos, que demonstrem segurança e desempenho clínico do algoritmo no contexto de uso pretendido.
Como tratar atualizações de algoritmo de IA após a aprovação?
Atualmente, qualquer alteração significativa em um SaMD aprovado exige nova submissão à ANVISA. O conceito de PCCP, Predetermined Change Control Plan, em discussão na revisão da RDC 657 permitiria ao fabricante descrever, antecipadamente, as mudanças esperadas no algoritmo e os critérios para validá-las sem precisar de uma nova aprovação a cada atualização, desde que dentro dos limites pré-definidos e aprovados.
Qual é a diferença entre SaMD e software de bem-estar (wellness)?
Software de bem-estar não possui finalidade médica declarada, monitora passos, sono ou nutrição sem vincular os dados a diagnóstico ou tratamento de condições de saúde. A linha divisória é a intended use (finalidade de uso): se o fabricante declara ou implica finalidade diagnóstica, terapêutica ou de monitoramento de condição médica, o software é SaMD e está sujeito à regulação. A ANVISA avalia tanto a bula/rotulagem quanto o material de marketing para determinar a finalidade real.
Uma startup sem CBPF pode vender SaMD Classe III no Brasil?
Não. O CBPF é pré-requisito para registro de dispositivos das classes III e IV. Uma startup pode, entretanto, contratar um fabricante parceiro que possua o CBPF, o chamado "modelo de contrato de fabricação", ou iniciar o processo de obtenção do certificado paralelamente ao desenvolvimento do produto. O CBPF é auditado pela ANVISA e tem validade de três anos, exigindo manutenção contínua do sistema de qualidade.
A regulação de SaMD no Brasil está em rápida evolução. A RDC 657/2022 estabeleceu as bases, mas a revisão em curso e o avanço do Marco Legal da IA indicam que o ambiente regulatório continuará se transformando nos próximos anos. Para engenheiros biomédicos e empreendedores de healthtech, dominar essa estrutura é uma vantagem competitiva real, e uma responsabilidade ética fundamental, dado o impacto direto desses produtos na segurança dos pacientes.
Para continuar aprofundando seu conhecimento em regulação e inovação em saúde, explore o Guia Definitivo de Engenharia Biomédica um recurso completo para profissionais e estudantes da área.
Referências e Recursos Externos
- ANVISA, Agência Nacional de Vigilância Sanitária: portal oficial com RDC 657/2022, guias de submissão e consultas públicas sobre SaMD.
- FDA, Medical Devices: banco de dados de dispositivos autorizados, guidance documents sobre IA/ML e o AI Action Plan.
- AAMI, Association for the Advancement of Medical Instrumentation: normas e recursos técnicos, incluindo publicações sobre IEC 62304 e cibersegurança em software médico.
Artigos relacionados
ANVISA e Dispositivos Médicos: Guia Completo de Regulamentação [Atualizado 2026]
Guia completo sobre regulamentação de dispositivos médicos pela ANVISA: classes de risco I-IV, fluxo de registro, documentação exigida, prazos, custos e as principais RDCs de 2022 a 2025. Conteúdo atualizado para profissionais de regulatory affairs, startups e fabricantes.
Certificação INMETRO para Equipamentos Médicos: Processo Passo a Passo
Guia completo sobre a certificação INMETRO de equipamentos eletromédicos no Brasil: Portaria 384/2020, normas IEC 60601, laboratórios acreditados, custos, prazos e o processo detalhado passo a passo para fabricantes e importadores.
Como Registrar um Dispositivo Médico na ANVISA: Roteiro Prático para Startups e Fabricantes
Guia completo do processo de registro de dispositivos médicos na ANVISA: classificação, CBPF, ensaios técnicos, submissão no sistema SOLICITA e acompanhamento. Prazos reais, custos atualizados e erros mais comuns a evitar.
IEC 60601: A Norma de Segurança de Equipamentos Eletromédicos Que Todo Engenheiro Precisa Conhecer
Entenda a estrutura completa da IEC 60601, o modelo sanduíche de normas gerais, colaterais e particulares, os conceitos de MOPP/MOOP, classificação de partes aplicadas e os requisitos de ensaio que definem a segurança de equipamentos eletromédicos no Brasil e no mundo.