A ISO 13485:2016 é a norma internacional que define os requisitos do Sistema de Gestão da Qualidade (SGQ) para fabricantes e fornecedores de dispositivos médicos, e desde 2 de fevereiro de 2026, passou a ser referência obrigatória também para o mercado norte-americano, após o FDA incorporar seus requisitos ao novo Quality Management System Regulation (QMSR). Para as mais de 1.200 empresas brasileiras do setor, que exportaram US$ 1,17 bilhão em 2024, entender e implementar corretamente essa norma é a diferença entre acessar ou perder mercados globais.
Este guia cobre cada cláusula da ISO 13485, traz checklists práticos de implementação, orienta sobre custos reais de certificação no Brasil e explica como integrar a norma com a RDC 665/2022 e o programa MDSAP. Para aprofundar a gestão de riscos exigida pela cláusula 7.1, consulte também nosso artigo sobre ISO 14971.
ISO 13485 x ISO 9001: Diferenças Fundamentais que Gestores Precisam Conhecer
A ISO 13485:2016 (3ª edição) é frequentemente confundida com a ISO 9001:2015, mas as diferenças são substanciais e impactam diretamente a estratégia de implementação. Enquanto a ISO 9001 adota a estrutura de alto nível Annex SL (10 cláusulas) e enfatiza a melhoria contínua a ISO 13485 mantém suas 8 cláusulas próprias e foca em manter a eficácia do SGQ não apenas melhorá-lo.
| Aspecto | ISO 13485:2016 | ISO 9001:2015 |
|---|---|---|
| Estrutura | 8 cláusulas (estrutura própria) | 10 cláusulas (Annex SL) |
| Foco | Manter eficácia do SGQ | Melhoria contínua |
| Rastreabilidade | Obrigatória e extensiva | Conforme aplicável |
| Ambiente limpo | Requisitos específicos (7.5.2) | Não abordado |
| Validação de software | Cláusula 4.1.6 explícita | Controle de mudanças genérico |
| Gestão de risco | Integrada em todo o produto | Abordagem baseada em risco geral |
| Registros | Vida útil do dispositivo | Período definido pela organização |
| Mercados-alvo | Dispositivos médicos (global) | Qualquer setor |
Em 2 de fevereiro de 2026, a FDA publicou a regra final (89 FR 7496) que substitui o antigo 21 CFR Part 820 pela QMSR, incorporando a ISO 13485:2016 por referência legal, conferindo à norma força de lei nos Estados Unidos.
— FDA, Quality Management System Regulation (QMSR), 89 FR 7496, 2026
Outro ponto crítico: a ISO 13485 exige entre 25 e 48 procedimentos documentados dependendo do escopo e complexidade dos produtos. Empresas que migram de um SGQ baseado em ISO 9001 geralmente precisam criar ou adaptar significativamente seus procedimentos de design e desenvolvimento, controle de produto não conforme, validação e rastreabilidade.
Estrutura da ISO 13485: As 8 Cláusulas que Regem o SGQ
Antes de iniciar a implementação, é fundamental compreender a lógica de cada cláusula e suas interdependências. A seguir, um mapa detalhado com os requisitos principais e os pontos de atenção mais frequentes em auditoria.
Cláusula 4, Sistema de Gestão da Qualidade
A Cláusula 4 estabelece os fundamentos do SGQ, incluindo o escopo, os requisitos gerais e os requisitos de documentação. O item 4.1.6 adicionado na versão 2016, trata especificamente da validação de software usado no SGQ, um dos requisitos mais frequentemente negligenciados em auditorias iniciais. Toda aplicação de software que afete a qualidade do produto deve ser validada antes do uso e revalidada após mudanças.
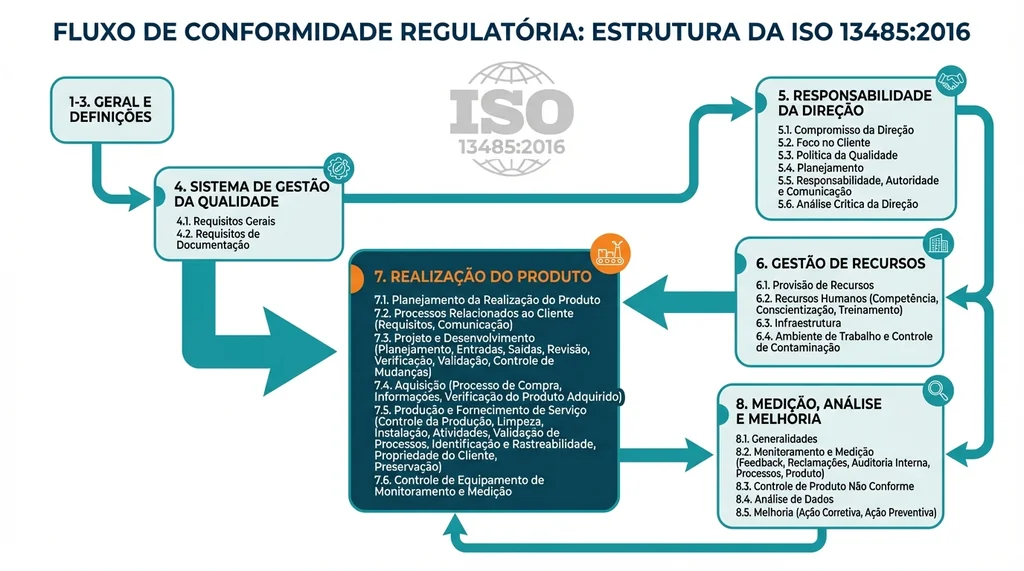
Descrição completa da imagem
Diagrama das 8 cláusulas da ISO 13485:2016 para sistemas de gestão da qualidade em dispositivos médicos, com destaque para cláusula 7.
O Manual da Qualidade (4.2.2) deve definir o escopo do SGQ, incluindo ou justificando exclusões, e descrever a interação entre os processos. Diferente da ISO 9001:2015, a ISO 13485 mantém o Manual como documento obrigatório.
Cláusula 5, Responsabilidade da Direção
A alta direção deve demonstrar comprometimento estabelecendo a política da qualidade, definindo objetivos mensuráveis, conduzindo análises críticas periódicas do SGQ e garantindo disponibilidade de recursos. Em auditorias de certificação, os auditores frequentemente entrevistam diretores para verificar se o compromisso é real ou apenas documental. A análise crítica pela direção (5.6) deve cobrir resultados de auditorias, feedback de clientes, desempenho de processos, status de ações corretivas e preventivas, além de mudanças que possam afetar o SGQ.
Cláusula 6, Gestão de Recursos
Abrange recursos humanos, infraestrutura e ambiente de trabalho. Para dispositivos médicos, o ambiente de trabalho (6.4) inclui requisitos específicos para controle de contaminação em produtos estéreis ou que necessitem de ambiente limpo. Há exigência de procedimentos documentados para controle de saúde de funcionários que possam afetar a qualidade do produto, um requisito frequentemente subestimado em implementações iniciais.
Cláusula 7, Realização do Produto (A Mais Crítica)
Com suas 14 subcláusulas, a Cláusula 7 é de longe a mais extensa e a que concentra o maior número de não-conformidades em auditorias. A subcláusula 7.3 (Projeto e Desenvolvimento) com 10 sub-itens, é especialmente crítica para fabricantes que desenvolvem seus próprios dispositivos.
| Subcláusula | Tema | Documentos Obrigatórios |
|---|---|---|
| 7.1 | Planejamento da realização do produto | Planos de qualidade, critérios de aceitação |
| 7.2 | Processos relacionados ao cliente | Requisitos do cliente, requisitos regulatórios |
| 7.3 | Projeto e Desenvolvimento | Plano P&D, inputs, outputs, revisões, verificação, validação, transferência, controle de mudanças, arquivo do DHF |
| 7.4 | Aquisição | Avaliação de fornecedores, especificações de compra |
| 7.5 | Produção e fornecimento de serviço | Instruções de trabalho, planos de controle, rastreabilidade, embalagem |
| 7.6 | Controle de equipamentos de monitoramento e medição | Registros de calibração, periodicidade |
O Device History File (DHF) ou Arquivo de Histórico do Dispositivo, deve conter evidências de que o projeto foi executado conforme o plano aprovado. O Device Master Record (DMR) é o conjunto de registros que define o produto finalizado: especificações, métodos de fabricação, procedimentos de controle de qualidade e embalagem. Ambos são verificados detalhadamente em auditorias de certificação e regulatórias.
Cláusula 8, Medição, Análise e Melhoria
Abrange monitoramento da satisfação do cliente, auditorias internas, monitoramento de processos e produtos, controle de produto não conforme, análise de dados, ações corretivas e preventivas (CAPA) e melhoria. O sistema de CAPA (Corrective and Preventive Action) é um dos processos mais auditados e frequentemente o mais problemático em implementações imaturas. A ISO 13485 exige investigação de causa raiz documentada, implementação de ações e verificação de eficácia.
Cronograma de Implementação: 12 a 18 Meses na Prática
A maioria das empresas leva entre 12 e 18 meses para implementar a ISO 13485 do zero. O tempo varia conforme o tamanho da empresa, complexidade dos produtos, maturidade do SGQ existente e disponibilidade de recursos dedicados. Abaixo, um cronograma realista dividido em quatro fases.
| Fase | Período | Atividades Principais | Entregáveis |
|---|---|---|---|
| 1, Diagnóstico e Planejamento | Meses 1-2 | Gap analysis, mapeamento de processos, definição do escopo, formação da equipe | Relatório de lacunas, plano de implementação, matriz de responsabilidades |
| 2, Desenvolvimento do SGQ | Meses 3-7 | Elaboração de procedimentos, instruções de trabalho, formulários; treinamento de equipes | Manual da Qualidade, 25-48 procedimentos documentados, registros de treinamento |
| 3, Implementação e Operação | Meses 8-12 | Operação do SGQ, coleta de evidências, auditorias internas, análise crítica pela direção | Registros de CAPA, relatórios de auditoria interna, ata de análise crítica |
| 4, Certificação | Meses 13-18 | Auditoria de estágio 1 (documentação), auditoria de estágio 2 (in loco), resolução de não-conformidades | Certificado ISO 13485, relatório de auditoria, plano de ações corretivas |
Checklist de Pré-Auditoria (Gap Analysis)
Antes de contratar um organismo certificador, realize uma análise de lacunas interna ou com consultoria especializada. Os pontos mais críticos a verificar:

- Documentação: Manual da Qualidade completo, todos os procedimentos obrigatórios redigidos e aprovados, sistema de controle de documentos operacional
- Projeto e Desenvolvimento (7.3): DHF completo para cada dispositivo no escopo, evidências de verificação e validação, controle de mudanças documentado
- Validação de Software (4.1.6): Inventário de softwares do SGQ, planos e relatórios de validação para cada sistema
- Gestão de Fornecedores (7.4): Lista aprovada de fornecedores críticos, critérios de avaliação documentados, acordos de qualidade com fornecedores de componentes críticos
- Rastreabilidade (7.5.9): Sistema capaz de identificar lotes, componentes e status de inspeção em todas as etapas
- CAPA (8.5.2/8.5.3): Pelo menos 3 ciclos completos de CAPA com investigação de causa raiz e verificação de eficácia documentados
- Auditoria Interna (8.2.4): Programa completo executado, todas as cláusulas auditadas, relatórios e ações corretivas registrados
- Gestão de Risco: Evidências de integração com ISO 14971 ao longo do ciclo de vida do produto
Custos Reais de Certificação no Brasil
O custo total de certificação ISO 13485 para empresas de pequeno e médio porte no Brasil varia entre R$ 80.000 e R$ 250.000 considerando todos os componentes. Abaixo, um detalhamento realista dos principais itens de custo.
| Item | Custo Estimado | Observação |
|---|---|---|
| Consultoria de implementação | R$ 40.000 – R$ 120.000 | Depende do escopo e maturidade do SGQ existente |
| Treinamentos (equipe interna) | R$ 8.000 – R$ 25.000 | Interpretação da norma, auditor interno, ferramentas de qualidade |
| Software de gestão do SGQ | R$ 5.000 – R$ 30.000/ano | eQMS como MasterControl, Qualio ou soluções nacionais |
| Calibração de equipamentos | R$ 3.000 – R$ 15.000 | Dependente do parque de equipamentos de medição |
| Auditoria de certificação (OC) | R$ 15.000 – R$ 45.000 | BSI, Bureau Veritas, SGS, TÜV, RINA, valores variam por OC |
| Auditorias de manutenção (anuais) | R$ 8.000 – R$ 20.000/ano | Obrigatórias para manutenção do certificado |
| Total implementação inicial | R$ 80.000 – R$ 250.000 | Empresas pequenas: extremo inferior; médias: extremo superior |
O ISO Survey 2024 registrou 31.215 certificados ISO 13485 válidos no mundo, cobrindo 43.957 sites, com tendência de crescimento impulsionada pela MDR europeia e pela QMSR americana.
— ISO Survey, 2024
Empresas que já possuem ISO 9001 certificada podem reduzir significativamente esses custos, tipicamente 30% a 40%, pois já possuem estrutura documental básica, cultura de auditorias e processos de CAPA estabelecidos. O principal esforço de migração concentra-se nas cláusulas específicas de dispositivos médicos: rastreabilidade extensiva, gestão de risco integrada, validação de processos e controles de projeto e desenvolvimento.
ISO 13485 e MDSAP: A Estratégia para Múltiplos Mercados
O Medical Device Single Audit Program (MDSAP) representa uma das maiores vantagens competitivas disponíveis para fabricantes brasileiros com ambições de exportação. Com um único ciclo de auditoria, a empresa obtém reconhecimento em cinco jurisdições: Brasil (ANVISA), Estados Unidos (FDA), Canadá (Health Canada), Austrália (TGA) e Japão (MHLW/PMDA).
A adoção do MDSAP no Brasil cresceu de forma expressiva: de apenas 4,7% das empresas em 2017 para 59,1% em 2023. Esse salto reflete tanto os incentivos regulatórios da ANVISA, que reconhece relatórios MDSAP como equivalente ao relatório de inspeção para fins de CBPF, quanto a crescente pressão dos mercados exportadores, especialmente o Canadá, que tornou o MDSAP obrigatório para fabricantes estrangeiros.
A estratégia ideal para empresas que almejam exportação é implementar a ISO 13485, buscar certificação inicial com um organismo credenciado pelo MDSAP, e usar o primeiro ciclo de auditoria MDSAP para consolidar o acesso simultâneo a múltiplos mercados. Para orientação sobre o cenário regulatório brasileiro, consulte nosso guia ANVISA de dispositivos médicos 2026.
ISO 13485 e a RDC 665/2022: Integração dos Sistemas
A RDC 665/2022 que estabelece as Boas Práticas de Fabricação (BPF) para dispositivos médicos no Brasil, é amplamente harmonizada com a ISO 13485. Isso significa que uma empresa devidamente certificada na norma internacional já atende a maior parte dos requisitos regulatórios da ANVISA, mas não todos.
As principais áreas de complementaridade e diferenças incluem:
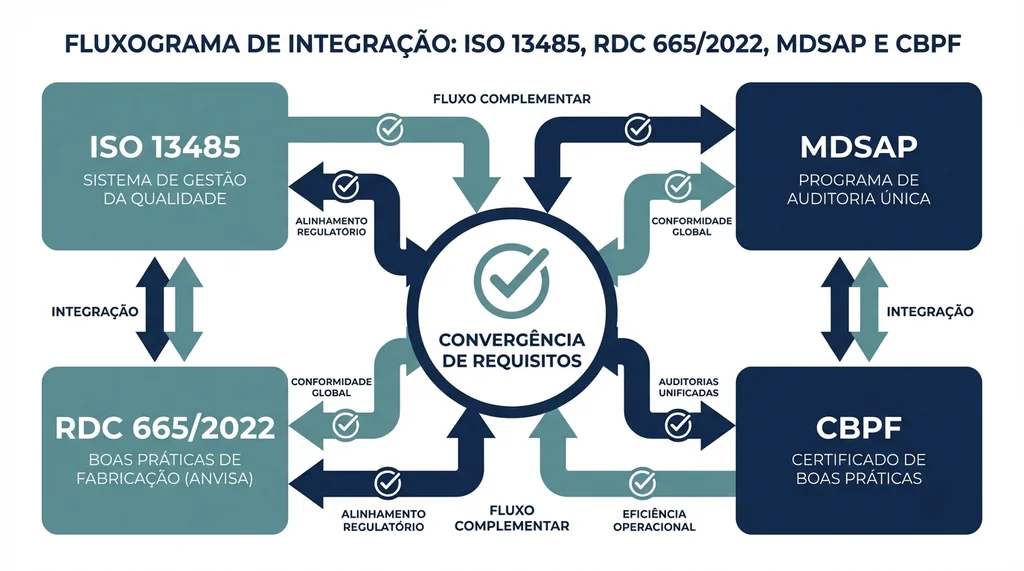
Descrição completa da imagem
Fluxograma de integração entre ISO 13485, RDC 665/2022 Boas Práticas de Fabricação, MDSAP e CBPF para dispositivos médicos no Brasil.
- Convergências: Controle de documentos e registros, gestão de fornecedores, rastreabilidade, controle de produto não conforme, CAPA, auditorias internas e análise crítica pela direção são praticamente idênticos entre as duas normas.
- Requisitos adicionais da RDC 665/2022: Notificação de eventos adversos à ANVISA, requisitos específicos para validação de processos de esterilização, e exigências de rotulagem em português conforme normas ABNT.
- CBPF (Certificado de Boas Práticas de Fabricação): Emitido pela ANVISA após inspeção. Para empresas com MDSAP, a ANVISA pode usar o relatório de auditoria MDSAP, reduzindo significativamente o esforço de inspeção.
O Brasil é o 14º maior mercado de dispositivos médicos do mundo e o maior da América Latina, com valor estimado em US$ 15,28 bilhões em 2024 e exportações de US$ 1,17 bilhão para 190 países, segundo dados do Relatório Setorial ABIMO 2024.
— ABIMO, Relatório Setorial, 2024
A recomendação prática para gestores é tratar a ISO 13485 como a espinha dorsal do SGQ e mapear os requisitos adicionais da RDC 665/2022 como complementos específicos do contexto regulatório brasileiro. Uma matriz de correlação entre cláusulas da norma e artigos da resolução é um documento de gestão essencial nesse processo.
Cláusula 7.3 em Detalhe: Projeto e Desenvolvimento
A Cláusula 7.3 é consistentemente a fonte do maior número de não-conformidades em auditorias ISO 13485, especialmente para fabricantes que desenvolvem produtos próprios. Seus 10 sub-itens cobrem todo o ciclo de vida do desenvolvimento:
7.3.1, Planejamento
Requer um plano de P&D documentado que defina as etapas do projeto, as revisões necessárias, as responsabilidades e as interfaces organizacionais e técnicas. O plano deve ser atualizado conforme o projeto evolui.
7.3.2 e 7.3.3, Entradas e Saídas
As entradas do projeto (7.3.2) devem incluir requisitos de desempenho e segurança, requisitos legais e regulatórios aplicáveis, informações de projetos anteriores similares e requisitos de gestão de risco. As saídas (7.3.3) devem ser verificáveis em relação às entradas e devem especificar características do produto críticas para sua segurança e uso correto.
7.3.4, 7.3.5 e 7.3.6, Revisão, Verificação e Validação
Esses três elementos formam o coração da garantia da qualidade no desenvolvimento. A revisão avalia se o projeto atende aos requisitos. A verificação confirma que as saídas do projeto atendem às entradas (o produto foi feito certo?). A validação confirma que o produto atende às necessidades do usuário e à aplicação pretendida (o produto certo foi feito?). Para softwares de dispositivos médicos, a IEC 62304 define os processos de ciclo de vida complementares.
7.3.9, Arquivo do Projeto (DHF)
O Device History File deve conter ou referenciar todos os registros de projeto e desenvolvimento, demonstrando que o processo foi executado conforme planejado. É a principal evidência auditada para verificar conformidade da cláusula 7.3 como um todo.
Preparando-se para a Próxima Edição da Norma
A ISO 13485:2025+ (nome provisório) está em desenvolvimento e deve ser publicada entre 2027 e 2028. As principais mudanças antecipadas incluem requisitos explícitos para dispositivos com componentes de Inteligência Artificial e Aprendizado de Máquina (AI/ML) diretrizes de cibersegurança para dispositivos conectados, maior alinhamento com o Regulamento de Dispositivos Médicos da União Europeia (MDR 2017/745) e integração de requisitos de sustentabilidade.
Para empresas que implementam ou renovam sua certificação agora, a recomendação é estruturar o SGQ de forma modular, documentando adequadamente os processos relacionados a software, conectividade e gerenciamento de dados, áreas que certamente serão endereçadas na próxima edição. Engenheiros biomédicos interessados em desenvolver competências nessa área podem verificar as certificações disponíveis para a área regulatória.
Perguntas Frequentes sobre ISO 13485
A ISO 13485 é obrigatória para vender dispositivos médicos no Brasil?
A ISO 13485 não é legalmente obrigatória no Brasil, mas a RDC 665/2022 exige que fabricantes de dispositivos médicos possuam um sistema de gestão da qualidade com requisitos equivalentes. Na prática, a certificação ISO 13485 é o caminho mais eficiente e reconhecido para demonstrar conformidade. Para exportação aos EUA, a partir de fevereiro de 2026, o QMSR do FDA incorpora os requisitos da ISO 13485 por referência, tornando-a essencial para o mercado norte-americano.
Qual a validade do certificado ISO 13485?
O certificado ISO 13485 tem validade de 3 anos, mas está condicionado à realização de auditorias de manutenção anuais (ou semestrais, dependendo do organismo certificador e do risco associado aos produtos). Uma não-conformidade grave identificada em auditoria de manutenção pode resultar em suspensão ou cancelamento do certificado antes do prazo de 3 anos.
Distribuidores e importadores precisam de ISO 13485?
Distribuidores e importadores que apenas comercializam produtos sem alterá-los podem implementar um escopo limitado do SGQ, excluindo as cláusulas de projeto e desenvolvimento (7.3) e partes da realização do produto. Porém, importadores que realizam atividades de rotulagem, embalagem ou armazenamento regulado têm escopo obrigatoriamente mais amplo. A ANVISA exige que importadores registrados possuam seus próprios controles de qualidade documentados.
Quanto tempo leva uma auditoria de certificação ISO 13485?
A duração da auditoria de certificação varia conforme o número de funcionários e a complexidade dos produtos, seguindo diretrizes de cálculo de dias de auditoria definidas pelo IAF (International Accreditation Forum). Para uma empresa de 50 a 200 funcionários com produtos de médio risco, uma auditoria típica de estágio 2 dura entre 4 e 8 dias de auditoria in loco, distribuídos por 2 auditores especializados. Empresas maiores ou com linha de produtos diversificada demandam mais tempo.
Como selecionar um organismo de certificação (OC) para ISO 13485?
O OC deve ser acreditado pelo INMETRO (para certificação com validade no Brasil) ou por um organismo de acreditação signatário do IAF MLA para o escopo de dispositivos médicos. Para certificação MDSAP, apenas organismos aprovados pelo programa são aceitos: BSI, Bureau Veritas, Dekra, SGS, TÜV SÜD e RINA são os principais atuando no Brasil. Solicite proposta de pelo menos três organismos, compare não apenas preço mas também experiência setorial específica com seus tipos de produtos e tempo de atendimento a respostas.
Para aprofundar sua compreensão do ecossistema regulatório de dispositivos médicos, explore nosso guia completo ANVISA 2026 e o artigo sobre gestão de riscos com ISO 14971 norma complementar e igualmente fundamental para qualquer fabricante de dispositivos médicos.
Referências e Recursos Oficiais
Artigos relacionados
ANVISA e Dispositivos Médicos: Guia Completo de Regulamentação [Atualizado 2026]
Guia completo sobre regulamentação de dispositivos médicos pela ANVISA: classes de risco I-IV, fluxo de registro, documentação exigida, prazos, custos e as principais RDCs de 2022 a 2025. Conteúdo atualizado para profissionais de regulatory affairs, startups e fabricantes.
Certificação INMETRO para Equipamentos Médicos: Processo Passo a Passo
Guia completo sobre a certificação INMETRO de equipamentos eletromédicos no Brasil: Portaria 384/2020, normas IEC 60601, laboratórios acreditados, custos, prazos e o processo detalhado passo a passo para fabricantes e importadores.
Como Registrar um Dispositivo Médico na ANVISA: Roteiro Prático para Startups e Fabricantes
Guia completo do processo de registro de dispositivos médicos na ANVISA: classificação, CBPF, ensaios técnicos, submissão no sistema SOLICITA e acompanhamento. Prazos reais, custos atualizados e erros mais comuns a evitar.
IEC 60601: A Norma de Segurança de Equipamentos Eletromédicos Que Todo Engenheiro Precisa Conhecer
Entenda a estrutura completa da IEC 60601, o modelo sanduíche de normas gerais, colaterais e particulares, os conceitos de MOPP/MOOP, classificação de partes aplicadas e os requisitos de ensaio que definem a segurança de equipamentos eletromédicos no Brasil e no mundo.