A ISO 14971 é o padrão internacional que define os requisitos para a gestão de risco de dispositivos médicos. Em sua terceira edição (2019), a norma estabelece um processo sistemático e documentado que os fabricantes devem seguir para identificar perigos, estimar e avaliar riscos, controlá-los e monitorar a eficácia dessas medidas ao longo de todo o ciclo de vida do produto. Reconhecida por reguladores como ANVISA, FDA, autoridades europeias (MDR/IVDR) e mais de 30 países a ISO 14971 é requisito fundamental para qualquer empresa que desenvolva, fabrique ou comercialize dispositivos médicos no mercado global.
Se você ainda não leu nosso guia sobre ISO 13485 e o sistema de gestão da qualidade para dispositivos médicos recomendamos começar por ele, a gestão de risco segundo a ISO 14971 é um subsistema crítico dentro do SGQ exigido pela ISO 13485. Neste artigo, vamos detalhar o processo completo, as ferramentas analíticas como FMEA e FTA, e como estruturar a documentação de rastreabilidade que os auditores e reguladores vão verificar.
O que é a ISO 14971 e por que ela é obrigatória?
A ISO 14971 não é uma norma de produto, ela não especifica qual nível de risco é aceitável para cada tipo de dispositivo. Em vez disso, ela define o processo que o fabricante deve seguir para tomar decisões fundamentadas sobre riscos, documentá-las e revisá-las continuamente. Isso é uma distinção fundamental: a norma exige que a empresa demonstre como chegou às suas conclusões, não apenas que o produto é seguro.
A evolução histórica da norma ilustra bem seu crescimento em escopo:
| Versão | Ano | Principal mudança |
|---|---|---|
| EN 1441 | 1997 | Primeira norma europeia de gestão de risco para DM |
| ISO 14971:2000 | 2000 | Primeira edição internacional |
| ISO 14971:2007 | 2007 | Expansão do escopo; reconhecimento global ampliado |
| ISO 14971:2019 | 2019 | Análise benefício-risco; cybersegurança; AFAP; "estado da arte" definido |
| Confirmação sem alterações | 2025 | Revisão periódica ISO concluída sem mudanças (março/2025) |
As principais mudanças da edição 2007 para a 2019 são relevantes para quem mantém sistemas legados: (1) a análise de risco-benefício foi invertida para benefício-risco colocando o benefício como ponto de partida da análise; (2) o conceito ALARP (As Low As Reasonably Practicable) foi substituído pelo AFAP (As Far As Possible), alinhando-se ao princípio europeu de que o fabricante deve reduzir o risco tanto quanto possível, sem o limitante "razoavelmente"; (3) o conceito de "dano" foi expandido para incluir dano a dados, propriedade e meio ambiente, além dos tradicionais danos físicos; (4) a cybersegurança foi explicitamente incluída no escopo; e (5) o conceito de "estado da arte" (state of the art) foi formalmente definido, impactando a determinação do risco residual aceitável.
O Regulamento de Dispositivos Médicos europeu (MDR 2017/745) menciona o termo "risco" 243 vezes em seu texto, contra apenas 69 vezes combinadas nas duas diretivas anteriores, evidenciando a centralidade da gestão de risco na regulamentação moderna.
— MDR 2017/745 (Regulamento UE), 2017
O Processo de Gestão de Risco segundo a ISO 14971
A norma organiza o processo em sete elementos principais, distribuídos nas cláusulas 4 a 10. É importante entender que não se trata de um fluxo linear, as atividades são iterativas e se retroalimentam ao longo do ciclo de vida do dispositivo.
Cláusula 4: Plano de Gestão de Risco
Antes de iniciar qualquer análise, o fabricante deve elaborar um Plano de Gestão de Risco específico para o dispositivo (ou família de dispositivos). O plano documenta: o escopo das atividades de gestão de risco, a atribuição de responsabilidades, os critérios de aceitabilidade de risco (incluindo critérios para riscos que serão avaliados individualmente e em conjunto), as atividades planejadas e as referências às normas e métodos que serão utilizados.
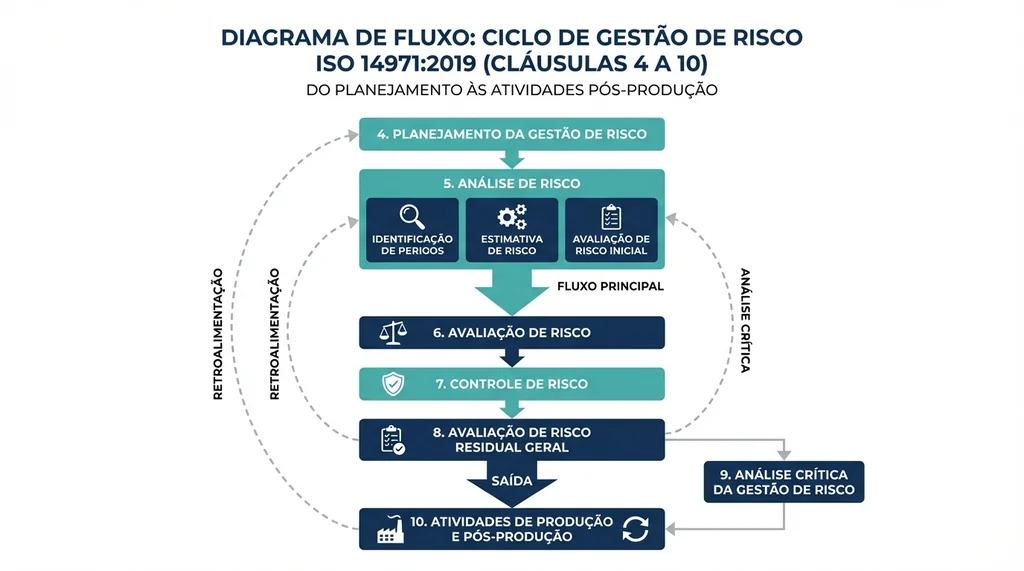
Descrição completa da imagem
Fluxograma do processo de gestão de risco ISO 14971:2019 com as cláusulas 4 a 10 e suas interconexões no ciclo de vida do dispositivo médico.
A definição dos critérios de aceitabilidade de risco no plano é uma das etapas mais críticas e frequentemente mal executadas. Muitas empresas copiam critérios genéricos de modelos, sem fundamentá-los em dados clínicos, literatura científica ou benchmarking com dispositivos similares. Os auditores do MDSAP (Medical Device Single Audit Program), cuja Tarefa 4 é dedicada à gestão de risco, e os revisores da ANVISA identificam isso rapidamente. Os critérios devem ser justificados e rastreáveis ao "estado da arte" da área médica em questão.
Cláusula 5: Análise de Risco
A análise de risco compreende três sub-etapas: (1) uso pretendido e identificação de uso indevido razoavelmente previsível, (2) identificação de características relacionadas à segurança, e (3) identificação de perigos e situações perigosas. A norma traz no Anexo C uma lista exemplificativa de questões a considerar, como características energéticas, biológicas, ambientais, de ergonomia e de software.
A estimativa de risco combina probabilidade e severidade. A probabilidade de ocorrência de dano é calculada como P = P1 × P2, onde P1 é a probabilidade de ocorrência da situação perigosa e P2 é a probabilidade de a situação perigosa resultar em dano. A severidade é classificada em escala que geralmente vai de desprezível (S1) a catastrófica (S5). A ISO 14971 não impõe uma escala específica, o fabricante define suas escalas no plano, mas deve justificá-las.
Cláusulas 6 e 7: Avaliação e Controle de Risco
Após estimar o risco, a empresa avalia se ele é aceitável conforme os critérios definidos no plano. Para riscos inaceitáveis (e, no espírito do AFAP, para todos os riscos), devem ser aplicadas medidas de controle em ordem de prioridade hierárquica estrita:
- Segurança inerente por design: eliminar o perigo ou reduzir o risco modificando o projeto (ex.: usar tensão elétrica mais baixa, eliminar arestas cortantes, usar material biocompatível).
- Medidas protetivas: proteções físicas, alarmes, sistemas de redundância, fusíveis, bloqueios de segurança.
- Informações para a segurança: rotulagem, instruções de uso (IFU), treinamento, apenas como último recurso, nunca como primeira linha de controle.
Essa hierarquia é inegociável. Um fabricante que tenta controlar um risco grave apenas com um aviso na IFU, sem avaliar medidas de design ou proteção, terá esse ponto questionado em qualquer auditoria regulatória, seja pela ANVISA conforme a RDC 751/2022 pelo FDA ou pelo organismo notificado europeu.
Cláusula 8: Avaliação do Risco Residual Global
Após a implementação e verificação das medidas de controle, o fabricante avalia o risco residual o risco que permanece após todos os controles. Se o risco residual individual for aceitável, a empresa deve então avaliar o risco residual global: a combinação de todos os riscos residuais do dispositivo. Aqui entra a análise de benefício-risco: se o risco residual global não for aceitável pelos critérios definidos, o fabricante deve demonstrar que os benefícios clínicos superam os riscos. Caso contrário, o dispositivo não pode ser comercializado.
Esta cláusula também exige que o fabricante determine quais informações sobre o risco residual devem ser incluídas nos materiais de comunicação com o usuário (IFU, rótulo, materiais de treinamento).
Cláusula 9: Revisão da Gestão de Risco
Antes de liberar o dispositivo para produção ou comercialização, o fabricante realiza uma revisão formal da gestão de risco para garantir que: o plano foi executado, todos os riscos foram avaliados, as medidas de controle foram verificadas, e a análise de benefício-risco global é favorável. Esta revisão é documentada no Relatório de Gestão de Risco.
Cláusula 10: Atividades de Produção e Pós-Produção
A ISO 14971:2019 expandiu significativamente os requisitos de monitoramento pós-comercialização em relação à edição 2007. O fabricante deve estabelecer um sistema para coletar e analisar informações de dispositivos em campo, reclamações, notificações de vigilância, literatura científica, dados de registros de saúde, e retroalimentar o processo de gestão de risco quando novas informações alteram a estimativa de riscos. Isso conecta diretamente a ISO 14971 ao sistema de vigilância pós-mercado exigido pela ANVISA e pelo Regulamento de Dispositivos Médicos europeu (MDR 2017/745).
A FDA autorizou um número recorde de 295 dispositivos com IA/ML em 2025, com mais de 1.200 acumulados até julho de 2025, dos quais 62% são Software as a Medical Device (SaMD) — reforçando a necessidade de processos robustos de gestão de risco pós-mercado.
— FDA, Artificial Intelligence and Machine Learning (AI/ML)-Enabled Medical Devices, 2025
Estimativa de Risco: Probabilidade e Severidade
A matriz de risco é a ferramenta mais visível do processo, mas sua correta calibração é frequentemente subestimada. Os cinco níveis de probabilidade típicos (adaptáveis pelo fabricante) são: Improvável (P1), Remoto (P2), Ocasional (P3), Provável (P4) e Frequente (P5). Os cinco níveis de severidade típicos são: Desprezível (S1, sem lesão ou lesão mínima), Menor (S2, lesão leve, reversível), Moderado (S3, lesão grave, reversível), Crítico (S4, lesão grave, irreversível) e Catastrófico (S5, morte).
| Probabilidade | S1 Desprezível | S2 Menor | S3 Moderado | S4 Crítico | S5 Catastrófico |
|---|---|---|---|---|---|
| P5 Frequente | Tolerável | Inaceitável | Inaceitável | Inaceitável | Inaceitável |
| P4 Provável | Aceitável | Tolerável | Inaceitável | Inaceitável | Inaceitável |
| P3 Ocasional | Aceitável | Aceitável | Tolerável | Inaceitável | Inaceitável |
| P2 Remoto | Aceitável | Aceitável | Aceitável | Tolerável | Inaceitável |
| P1 Improvável | Aceitável | Aceitável | Aceitável | Aceitável | Tolerável |
Riscos classificados como "Toleráveis" devem ser reduzidos tanto quanto possível (princípio AFAP) e documentar por que não foi possível reduzi-los mais. Riscos "Inaceitáveis" exigem medidas de controle obrigatórias. Apenas riscos "Aceitáveis" podem ser mantidos sem ação adicional, mas ainda devem ser documentados no arquivo de gestão de risco.
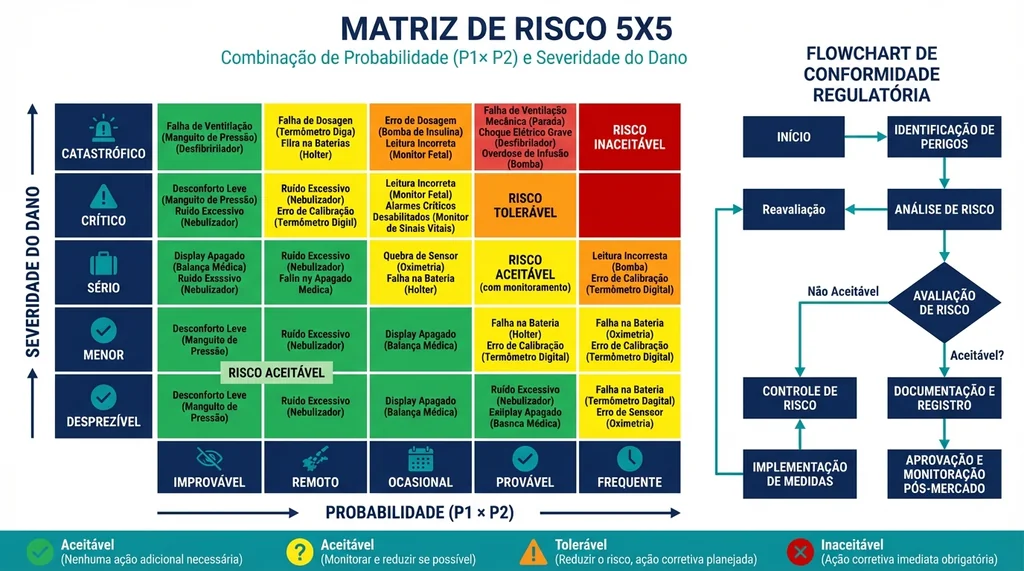
Descrição completa da imagem
Matriz de risco 5x5 da ISO 14971 com níveis de probabilidade e severidade para classificação de riscos em dispositivos médicos segundo critérios ALAP/AFAP.
Ferramentas de Análise de Risco: FMEA, FTA e HAZOP
A ISO 14971 não prescreve ferramentas específicas de análise, o fabricante escolhe as mais adequadas ao tipo de dispositivo e ao nível de maturidade do projeto. O guia complementar ISO/TR 24971:2020 traz orientações detalhadas e exemplos de aplicação de cada ferramenta.
FMEA (Failure Mode and Effects Analysis)
O FMEA é a ferramenta mais amplamente utilizada na indústria de dispositivos médicos. Parte de uma abordagem bottom-up: para cada componente ou função do dispositivo, o analista identifica os modos de falha possíveis, seus efeitos sobre o usuário/paciente, as causas da falha e as medidas de controle existentes. O resultado é tipicamente apresentado em uma planilha com colunas padronizadas:
Em 2024, os recalls de dispositivos médicos nos EUA atingiram 1.059 eventos — o maior nível em quatro anos — com falha do dispositivo como causa líder e problemas de software como segunda causa mais comum.
— FDA, Medical Device Recalls Database, 2024
| Coluna FMEA | Descrição | Exemplo (bomba de infusão) |
|---|---|---|
| Função/Item | Componente ou função analisada | Válvula de oclusão |
| Modo de falha | Como o item pode falhar | Válvula não fecha completamente |
| Efeito da falha | Consequência para o paciente/usuário | Sobredosagem do medicamento |
| Causa da falha | Por que o modo de falha ocorre | Desgaste do elastômero; partículas no fluido |
| Severidade (S) | Nível de dano potencial | S4, Crítico (lesão grave) |
| Probabilidade (P) | P1 × P2 estimada | P2, Remoto |
| Risco inicial | Classificação antes dos controles | Tolerável |
| Medida de controle | Ação de mitigação implementada | Alarme de oclusão; filtro no tubo de entrada |
| Risco residual | Classificação após os controles | Aceitável |
O FMEA pode ser aplicado no nível de sistema (sFMEA), subsistema, componente ou processo (pFMEA). Para dispositivos que envolvem software como dispositivo médico (SaMD) o FMEA de software deve ser integrado à classificação de segurança de software segundo a IEC 62304 classes A, B e C correspondem a diferentes níveis de rigor de desenvolvimento e testes, diretamente vinculados à severidade dos riscos identificados na ISO 14971.
FTA (Fault Tree Analysis)
O FTA é uma abordagem top-down: parte de um evento indesejado de alto nível (ex.: "paciente recebe dose excessiva") e decompõe as causas em uma árvore lógica de eventos intermediários e básicos, usando portas AND/OR. É especialmente útil para analisar sistemas complexos com múltiplos caminhos de falha e interações entre subsistemas eletrônicos, mecânicos e de software. O FTA permite calcular a probabilidade do evento de topo quando as probabilidades dos eventos básicos são conhecidas ou estimáveis.
HAZOP e PHA
O HAZOP (Hazard and Operability Study) utiliza palavras-guia (como "mais", "menos", "nenhum", "inverso") aplicadas a parâmetros de processo para identificar desvios e seus efeitos. É amplamente usado em dispositivos com fluxo de fluidos, como equipamentos de diálise, sistemas de anestesia e ventiladores mecânicos. O PHA (Preliminary Hazard Analysis) é aplicado nas fases iniciais do desenvolvimento, quando o design ainda não está detalhado, para identificar perigos de alto nível e orientar decisões arquiteturais.
ISO 14971 na Prática: O Arquivo de Gestão de Risco
O Arquivo de Gestão de Risco (Risk Management File, RMF) é o conjunto de documentos e registros que evidencia a execução completa do processo. Não é um único documento, mas uma coleção rastreável que inclui:
- Plano de Gestão de Risco (com critérios de aceitabilidade justificados)
- Análise de uso pretendido e características relacionadas à segurança
- Registros de identificação de perigos e situações perigosas
- Estimativas de risco (pré-controle) com justificativas de probabilidade e severidade
- Planilhas FMEA / FTA / outros relatórios de análise
- Evidências de implementação e verificação das medidas de controle
- Avaliação do risco residual e análise de benefício-risco global
- Relatório de Gestão de Risco (resumo executivo da revisão, Cláusula 9)
- Registros de monitoramento pós-produção e ações tomadas (Cláusula 10)
A rastreabilidade bidirecional é um requisito crítico: cada risco identificado deve ser rastreável às medidas de controle implementadas, e cada medida de controle deve ser rastreável ao risco que mitiga. Ferramentas de ALM (Application Lifecycle Management) como Jama Connect, Polarion ou mesmo planilhas Excel bem estruturadas podem ser usadas, o importante é que o auditor consiga navegar do perigo ao controle e vice-versa sem lacunas.

Integração com Requisitos Regulatórios Globais
Um dos maiores ativos da ISO 14971 é seu reconhecimento global, que permite ao fabricante usar um único processo de gestão de risco para atender a múltiplos mercados:
| Regulador / Marco regulatório | Como a ISO 14971 é referenciada | Observações |
|---|---|---|
| ANVISA (Brasil) | RDC 751/2022 e normativas complementares | Exigida para registro e renovação de DM; ver guia RDC 751 |
| FDA (EUA) | Reconhecida como consensus standard (FDA Recognition) | 510(k), PMA e De Novo referenciam a norma |
| CE (Europa) | Harmonizada sob MDR 2017/745 e IVDR 2017/746 | EN ISO 14971:2019 é norma harmonizada vigente |
| Health Canada | Referenciada no Medical Devices Regulations | Parte do MDSAP (Canada, EUA, Brasil, Japão, Austrália) |
| MDSAP | Tarefa 4 dedicada à gestão de risco | Auditoria única aceita por 5 países |
Para o mercado brasileiro, a ANVISA exige que o dossiê técnico de registro inclua evidência de que a gestão de risco foi conduzida conforme a ISO 14971. A ausência ou insuficiência do arquivo de gestão de risco é uma das principais causas de exigência (pedido de complementação) nos processos de registro de dispositivos médicos de alto risco (Classes III e IV).
Para dispositivos com componente de software, a integração com a IEC 62304 é mandatória: a classificação de segurança do software (A, B ou C) deve ser determinada com base nos riscos identificados segundo a ISO 14971, e as atividades de desenvolvimento e verificação de software devem ser proporcionais a essa classificação.
Cybersegurança e Tendências Futuras
A ISO 14971:2019 foi a primeira edição a incluir explicitamente a cybersegurança no escopo de gestão de risco. Vulnerabilidades de software e de conectividade (rede hospitalar, Bluetooth, nuvem) são tratadas como fontes de perigo que devem ser analisadas pelo mesmo processo, identificação de perigo, estimativa de risco, controle e monitoramento. A norma IEC 81001-5-1:2021 (Atividades de ciclo de vida de segurança para software de saúde, cybersegurança) é o documento complementar específico para ameaças cibernéticas, e sua aplicação é crescentemente exigida por reguladores europeus e norte-americanos.
A revisão periódica de março de 2025 confirmou a ISO 14971:2019 sem alterações, mas os grupos de trabalho do ISO/TC 210 já estão avaliando como futuras revisões abordarão IA/ML em dispositivos médicos, área onde a gestão de risco enfrenta desafios inéditos relacionados à imprevisibilidade de algoritmos adaptativos e à necessidade de monitoramento contínuo de performance em campo. O documento de orientação ISO/TR 24971:2020 permanece como a principal referência de apoio à implementação prática da norma.
Erros Comuns na Implementação da ISO 14971
Com base em padrões frequentes de não-conformidade identificados em auditorias MDSAP, ISO 13485 e revisões regulatórias, os erros mais recorrentes são:
- Critérios de aceitabilidade sem justificativa: tabelas de risco copiadas de modelos genéricos sem fundamentação em dados clínicos ou "estado da arte".
- FMEA desatualizado: planilhas criadas durante o desenvolvimento e nunca revisadas após mudanças de design, reclamações de campo ou novas evidências da literatura.
- Falta de rastreabilidade: medidas de controle listadas no FMEA sem evidência de implementação verificada (registros de teste, inspeção ou qualificação).
- Análise de benefício-risco ausente: a empresa avalia riscos individuais mas não documenta a análise do risco residual global e a comparação com benefícios clínicos.
- Desconexão com o pós-mercado: o arquivo de gestão de risco é fechado no lançamento do produto e nunca alimentado com dados de vigilância pós-mercado, reclamações ou recalls de concorrentes.
- Hierarquia de controle não respeitada: riscos graves controlados apenas com advertências na IFU, sem avaliação de alternativas de design ou proteção.
- Software sem integração com IEC 62304: classificação de segurança do software não rastreada aos riscos ISO 14971, ou vice-versa.
FAQ, Perguntas Frequentes sobre ISO 14971
A ISO 14971 se aplica a acessórios e peças de reposição?
Sim. A norma se aplica a dispositivos médicos conforme definido pelas regulamentações aplicáveis, e na maioria dos marcos regulatórios, acessórios e peças de reposição destinados a uso com dispositivos médicos são eles próprios classificados como dispositivos médicos. O fabricante deve conduzir a gestão de risco para o acessório de forma independente, mas também deve considerar os riscos introduzidos pela combinação do acessório com o dispositivo principal.
Qual a diferença entre FMEA de produto e FMEA de processo?
O FMEA de produto (dFMEA, design FMEA) analisa modos de falha relacionados ao design do dispositivo e seus efeitos sobre o usuário/paciente. O FMEA de processo (pFMEA) analisa modos de falha nos processos de fabricação e como esses podem resultar em defeitos no produto acabado que causem dano. Ambos são relevantes para a ISO 14971, mas o dFMEA é o mais diretamente vinculado ao processo de gestão de risco do produto. O pFMEA conecta-se mais ao controle de processo no contexto da ISO 13485.
Como a ISO 14971 trata riscos de dispositivos combinados (drug-device combinations)?
Combinações de produto (drug-device combinations, como autoaplicadores de insulina ou stents farmacológicos) exigem que o fabricante considere a interação entre o componente de dispositivo e o componente farmacológico como fonte adicional de perigos. A ISO 14971 se aplica ao componente de dispositivo; a gestão de risco do componente farmacológico segue normativas próprias (ICH Q9, por exemplo). O Relatório de Gestão de Risco deve documentar explicitamente como a interação entre os componentes foi avaliada.
É necessário contratar uma consultoria especializada para implementar a ISO 14971?
Não é obrigatório, mas é altamente recomendável para empresas que iniciam o processo ou que enfrentaram não-conformidades em auditorias anteriores. A norma exige competência técnica para definir critérios de aceitabilidade, conduzir análises de FMEA e FTA, e integrar o processo com os demais requisitos do SGQ. Internamente, o time deve incluir profissionais com formação em engenharia (biomedical, mecânica, elétrica, de software), regulatory affairs e qualidade, com experiência no tipo de dispositivo em questão.
O Relatório de Gestão de Risco é o mesmo que o Arquivo de Gestão de Risco?
Não. O Arquivo de Gestão de Risco (Risk Management File) é o conjunto completo de todos os documentos e registros do processo, análises, evidências de controle, registros de pós-mercado, etc. O Relatório de Gestão de Risco (Risk Management Report) é um documento específico gerado na Cláusula 9, que resume os resultados da revisão final antes do lançamento: confirma que o plano foi executado, que os critérios de aceitabilidade foram atendidos e que a análise de benefício-risco global é favorável. É essencialmente o "atestado de conclusão" do processo para aquele ciclo de desenvolvimento.
Para aprofundar sua compreensão do ecossistema regulatório brasileiro, leia também nossos guias sobre o sistema de gestão da qualidade ISO 13485 o processo de registro na ANVISA e a classificação de dispositivos médicos pela RDC 751/2022.
Artigos relacionados
ANVISA e Dispositivos Médicos: Guia Completo de Regulamentação [Atualizado 2026]
Guia completo sobre regulamentação de dispositivos médicos pela ANVISA: classes de risco I-IV, fluxo de registro, documentação exigida, prazos, custos e as principais RDCs de 2022 a 2025. Conteúdo atualizado para profissionais de regulatory affairs, startups e fabricantes.
Certificação INMETRO para Equipamentos Médicos: Processo Passo a Passo
Guia completo sobre a certificação INMETRO de equipamentos eletromédicos no Brasil: Portaria 384/2020, normas IEC 60601, laboratórios acreditados, custos, prazos e o processo detalhado passo a passo para fabricantes e importadores.
Como Registrar um Dispositivo Médico na ANVISA: Roteiro Prático para Startups e Fabricantes
Guia completo do processo de registro de dispositivos médicos na ANVISA: classificação, CBPF, ensaios técnicos, submissão no sistema SOLICITA e acompanhamento. Prazos reais, custos atualizados e erros mais comuns a evitar.
IEC 60601: A Norma de Segurança de Equipamentos Eletromédicos Que Todo Engenheiro Precisa Conhecer
Entenda a estrutura completa da IEC 60601, o modelo sanduíche de normas gerais, colaterais e particulares, os conceitos de MOPP/MOOP, classificação de partes aplicadas e os requisitos de ensaio que definem a segurança de equipamentos eletromédicos no Brasil e no mundo.