A tecnovigilância é o conjunto de ações de vigilância pós-mercado aplicadas a produtos de saúde, equipamentos médicos, implantes, kits diagnósticos e correlatos, com o objetivo de identificar, avaliar e prevenir eventos adversos e queixas técnicas. No Brasil, esse sistema é coordenado pela ANVISA e operacionalizado por meio do NOTIVISA sendo de cumprimento obrigatório para fabricantes, importadores e detentores de registro. Compreender as obrigações legais, os prazos de notificação e as boas práticas desse sistema é indispensável para manter a conformidade regulatória e, sobretudo, proteger pacientes e profissionais de saúde.
Se você ainda está estruturando a base regulatória da sua empresa, consulte também nosso guia completo sobre dispositivos médicos na ANVISA em 2026 e o artigo sobre como registrar um dispositivo médico na ANVISA. Para a dimensão de gestão de riscos que antecede a vigilância pós-mercado, veja nossa análise da ISO 14971 para gestão de risco.
O que é Tecnovigilância e por que ela é obrigatória
A tecnovigilância brasileira tem sua norma central na RDC 67/2009 ainda em vigor em 2026. Ela define o sistema de vigilância pós-comercialização de produtos de saúde sujeitos ao regime de vigilância sanitária, abrangendo três categorias principais de ocorrências:
- Eventos adversos: danos à saúde de paciente, usuário ou terceiro, relacionados ao uso de produto de saúde.
- Queixas técnicas: desvios de qualidade que não chegaram a causar dano, mas que poderiam ter causado ou que comprometem as características do produto.
- Desvios de qualidade: inconformidades detectadas antes do uso, durante a fabricação ou distribuição.
A obrigatoriedade não se limita apenas a notificar eventos já ocorridos. A legislação exige que fabricantes e importadores mantenham um sistema estruturado de vigilância pós-mercado com procedimentos documentados para coleta, análise e resposta a informações de campo. Essa exigência foi reforçada pela RDC 848/2024 que ampliou os requisitos de avaliação clínica periódica e introduziu critérios específicos para dispositivos com conectividade (cibersegurança) e nanomateriais.
Estrutura do Sistema de Tecnovigilância: Três Níveis
O sistema brasileiro opera em três níveis hierárquicos complementares:
Nível Federal: ANVISA, GGMON e GETEC
A ANVISA é a autoridade regulatória central. Dentro dela, a GGMON (Gerência-Geral de Monitoramento de Mercado) coordena as ações nacionais de pós-mercado. A GETEC (Gerência de Tecnovigilância) é responsável especificamente pela análise das notificações de produtos médicos, emissão de alertas e investigação de eventos graves. O SISTEC sistema de alertas da ANVISA, acumula mais de 5.060 alertas emitidos desde sua criação, incluindo proibições de produtos e recalls.
Nível Estadual e Municipal: as VISAs
As Vigilâncias Sanitárias (VISAs) estaduais e municipais são o braço executivo regional. Elas recebem notificações locais, realizam inspeções em estabelecimentos e podem determinar medidas cautelares imediatas, como a suspensão de uso de um equipamento, enquanto a ANVISA conduz a investigação federal. A integração entre os três níveis é essencial para a velocidade de resposta a eventos graves.
Rede Sentinela: os Olhos do Sistema
A Rede Sentinela é composta por hospitais e serviços de saúde de referência que atuam como notificadores qualificados e vigilantes ativos. Em dezembro de 2025, a rede contava com 281 unidades em todos os 27 estados. A RDC 872/2024 modernizou o marco regulatório da Rede Sentinela, reforçando os critérios de adesão, as responsabilidades dos serviços e os mecanismos de retroalimentação de informações. Engenheiros clínicos nesses hospitais são figuras centrais na identificação e registro de ocorrências, veja mais em nosso artigo sobre manutenção de equipamentos hospitalares.
Em 2025, a Rede Sentinela respondeu por aproximadamente 25% das notificações de tecnovigilância no Brasil, originárias de 162 das 281 instituições credenciadas em todas as unidades federativas.
— ANVISA, Boletim Rede Sentinela Vol. 2, 2025
NOTIVISA: O Sistema Eletrônico de Notificação
O NOTIVISA (Sistema de Notificações em Vigilância Sanitária) é a plataforma eletrônica oficial da ANVISA para registro de eventos adversos e queixas técnicas, em operação desde 2006. O acesso é feito pelo portal da ANVISA, com cadastro por CPF/CNPJ, e há módulos distintos para:
- Fabricantes e importadores (notificadores obrigatórios)
- Serviços de saúde e Rede Sentinela
- Profissionais de saúde
- Cidadãos (via e-NOTIVISA plataforma experimental que permite comunicação direta entre cidadão e empresa)
Uma notificação no NOTIVISA deve conter, no mínimo: identificação do produto (nome, número de lote, registro ANVISA), descrição detalhada da ocorrência, dados do paciente/usuário (anonimizados), data do evento e ações imediatas já tomadas. Após o envio, a empresa recebe um número de protocolo e pode acompanhar o andamento pelo sistema.
Prazos de Notificação: o que a Lei Exige
Os prazos são um dos pontos mais críticos da conformidade. A RDC 67/2009 estabelece três categorias de urgência:
| Tipo de Ocorrência | Prazo para Notificação Inicial | Prazo para Relatório Final |
|---|---|---|
| Morte ou grave ameaça à saúde pública | 72 horas | 30 dias após a notificação inicial |
| Evento adverso grave (hospitalização, dano permanente) | 10 dias úteis | 30 dias após a notificação inicial |
| Mau funcionamento sem dano imediato | 30 dias corridos | Conforme investigação |
O prazo de 72 horas conta a partir do momento em que a empresa toma conhecimento do evento, não da data do evento em si. Por isso, é fundamental que o sistema de Reclamações e SAC da empresa esteja integrado ao processo de tecnovigilância, com triagem imediata de ocorrências críticas. Atrasos nas notificações são infrações sanitárias sujeitas a penalidades, independentemente de dolo.
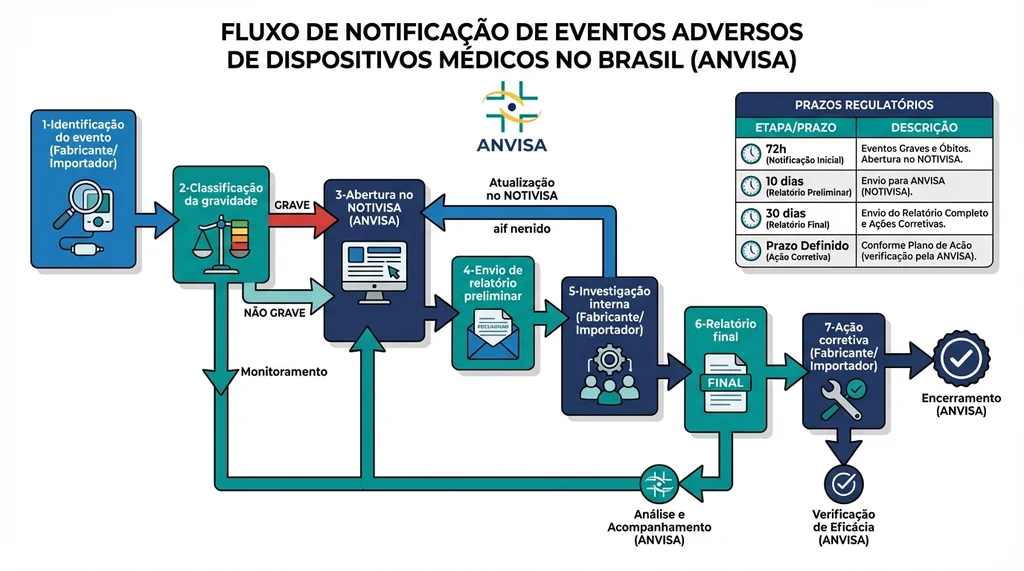
Descrição completa da imagem
Fluxograma do processo de notificação de eventos adversos de dispositivos médicos no sistema NOTIVISA da ANVISA com prazos regulatórios.
Ações de Campo: Recalls e Correções sob a RDC 551/2021
A RDC 551/2021 substituiu a RDC 23/2012 e consolidou o marco regulatório para ações de campo de produtos de saúde no Brasil. Uma ação de campo é qualquer medida tomada pelo detentor de registro para corrigir ou remover um produto que apresenta risco à saúde ou que está em desacordo com a legislação sanitária.
Tipos de Ação de Campo
| Tipo | Descrição | Produto permanece no mercado? |
|---|---|---|
| Recall | Recolhimento do produto já distribuído ou em uso | Não (temporariamente) |
| Correção de Campo | Modificação, ajuste, reparo ou reetiquetagem in loco | Sim, após correção |
| Suspensão de Uso | Instrução para interromper o uso até nova avaliação | Suspenso |
| Retirada do Mercado | Remoção definitiva de produtos não vendidos | Não |
| Notificação a Clientes | Comunicação de risco sem recolhimento físico | Sim, com comunicado |
A empresa deve submeter à ANVISA um Plano de Ação de Campo com cronograma, lista de lotes afetados, estratégia de comunicação e mecanismo de rastreamento. A ANVISA avalia o plano e pode exigir ajustes. O detentor de registro é responsável por comunicar distribuidores, hospitais e usuários finais, além de documentar cada unidade recuperada ou corrigida.
Classificação de Risco para Ações de Campo
Seguindo as diretrizes do IMDRF (do qual o Brasil é membro fundador), as ações de campo são classificadas por nível de risco:
- Classe I: situação em que existe probabilidade razoável de que o uso ou exposição ao produto cause danos graves à saúde ou morte.
- Classe II: situação em que o uso ou exposição pode causar danos temporários à saúde, mas sem probabilidade de serem graves ou irreversíveis.
- Classe III: situação em que o uso ou exposição não deve causar danos à saúde.
Penalidades pelo Descumprimento
O descumprimento das obrigações de tecnovigilância configura infração sanitária, sujeita às penalidades previstas na Lei 6.437/1977 e no Decreto 8.077/2013. As sanções incluem:
- Advertência por escrito
- Apreensão e inutilização de produtos
- Suspensão de venda, distribuição ou uso
- Cancelamento do registro do produto
- Proibição de fabricação ou importação
- Multas de R$ 2.000 a R$ 1.500.000 por infração
- Interdição parcial ou total do estabelecimento
Exemplos recentes ilustram a severidade da aplicação: em fevereiro de 2026, a ANVISA determinou a proibição de produtos de empresas como TMA Medicina, Alfalagos e Capilar Brasil por descumprimento de normas regulatórias, com alertas publicados no SISTEC e ampla divulgação pública. A reputação da empresa junto a compradores hospitalares e importadores internacionais também é afetada significativamente por registros de ações coercitivas.
As multas por infrações sanitárias relacionadas a tecnovigilância variam de R$ 2.000 (infrações leves) a R$ 1.500.000 (infrações gravíssimas), com valores duplicados em caso de reincidência, conforme a Lei 6.437/1977.
— Lei 6.437/1977 (Lei de Infrações Sanitárias), Brasil

Boas Práticas para Fabricantes e Importadores
Cumprir a legislação é o mínimo. As empresas com programas maduros de tecnovigilância vão além e implementam um sistema robusto de Post-Market Surveillance (PMS) integrado ao ciclo de vida do produto. As práticas a seguir são reconhecidas pela ANVISA e estão alinhadas com os padrões do IMDRF:
1. Procedimento Operacional Padrão (POP) de Tecnovigilância
Documente em POP o fluxo completo: como uma reclamação entra na empresa (SAC, e-mail, visita técnica), como é triada quanto à gravidade, quem decide sobre a necessidade de notificação, quem abre o NOTIVISA e quem assina o relatório final. O POP deve ser testado com simulações periódicas e atualizado a cada mudança regulatória.
2. Treinamento Contínuo da Equipe
Todos os funcionários que interagem com informações de campo, vendedores, assistência técnica, SAC, qualidade, devem receber treinamento específico sobre tecnovigilância. A identificação precoce de sinais de alerta depende da capacidade de reconhecer uma ocorrência relevante antes que ela escale.
3. Rastreabilidade Lote a Lote
Manter registros de distribuição por lote, quais hospitais, distribuidores e regiões receberam cada lote, é indispensável para uma ação de campo eficaz. Nesse contexto, vale acompanhar as novidades sobre o sistema de rastreamento e identificação de dispositivos médicos aprovado pela ANVISA. Sem essa rastreabilidade, um recall de Classe I pode se tornar uma operação caótica, com risco de unidades afetadas continuarem em uso. A ISO 14971 e os requisitos de sistema de gestão de qualidade (ISO 13485) fornecem a estrutura de base.
4. Monitoramento Proativo da Literatura e Bases de Dados
Um programa de PMS inclui monitoramento sistemático de bancos de dados públicos de eventos adversos, como o MAUDE da FDA e o EUDAMED para identificar sinais de segurança emergentes em produtos similares ao seu, mesmo antes de receber reclamações diretas. Essa prática é exigida formalmente pela regulamentação europeia e é uma boa prática reconhecida pela ANVISA.
5. Revisão Periódica do Relatório de PMS
A RDC 848/2024 exige que fabricantes de dispositivos de maior risco mantenham relatórios de avaliação clínica com revisão periódica, incorporando os dados de pós-mercado coletados. Esse relatório deve demonstrar que o balanço risco-benefício do produto permanece favorável à luz das evidências acumuladas.
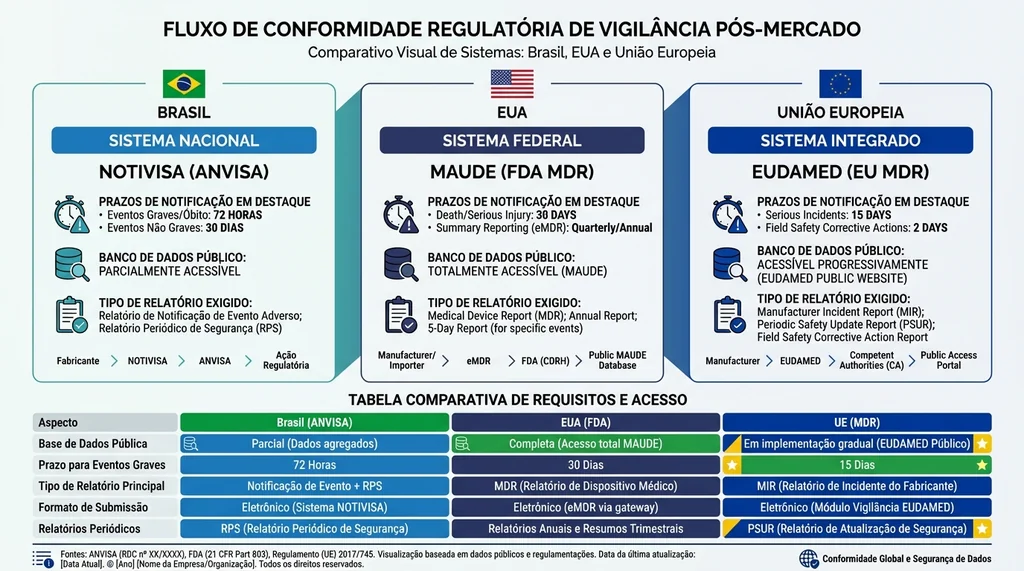
Tecnovigilância Global: Comparação com FDA e EU MDR
O sistema brasileiro de tecnovigilância está em processo de crescente harmonização com os padrões internacionais, sobretudo por meio da participação do Brasil no IMDRF e pela adoção do MDSAP (Medical Device Single Audit Program). Veja como os três principais sistemas se comparam:
| Aspecto | Brasil (ANVISA) | EUA (FDA) | União Europeia (EU MDR) |
|---|---|---|---|
| Norma principal | RDC 67/2009 + RDC 848/2024 | 21 CFR Part 803 (MDR) | EU MDR 2017/745 |
| Sistema de notificação | NOTIVISA | FDA MedWatch / MAUDE | EUDAMED |
| Prazo para morte/evento grave | 72h / 10 dias | 30 dias (5 dias para MDR imediato) | 15 dias (2 dias para grave) |
| Base de dados pública | SISTEC (alertas) | MAUDE (público) | EUDAMED (em implantação) |
| Recalls | RDC 551/2021 | 21 CFR Part 806 | Art. 83-89 EU MDR |
| PMS periódico obrigatório | Sim (RDC 848/2024) | Sim (MDR, PMSR) | Sim (PMSR/PSUR) |
A principal diferença está nos prazos: o sistema europeu é o mais exigente para eventos imediatos (2 dias para situações com risco de morte iminente), enquanto o brasileiro é mais ágil que o americano para as categorias mais graves (72h vs. 30 dias do FDA geral). Para empresas que exportam para múltiplos mercados, o alinhamento com os guias do IMDRF é a estratégia mais eficiente para atender simultaneamente às exigências de cada jurisdição.
O FDA MAUDE recebeu mais de 2 milhões de relatórios de dispositivos médicos (MDRs) anuais em 2024, um aumento de 5 vezes desde 2011, evidenciando a escala do sistema americano de vigilância pós-mercado frente ao brasileiro.
— FDA, MAUDE Database Statistics, 2024
Perguntas Frequentes sobre Tecnovigilância
Quem é obrigado a notificar eventos adversos à ANVISA?
Todo fabricante, importador e detentor de registro de produto de saúde sujeito à vigilância sanitária é obrigado a notificar eventos adversos e queixas técnicas relevantes. Serviços de saúde integrantes da Rede Sentinela também têm obrigação formal de notificação. Profissionais de saúde e cidadãos podem notificar voluntariamente pelo NOTIVISA ou pelo e-NOTIVISA.
O que acontece se meu produto causar um evento adverso e eu não notificar?
O não cumprimento do dever de notificação configura infração sanitária autônoma, independentemente de investigação sobre o evento em si. As penalidades vão de advertência a multas de até R$ 1,5 milhão e cancelamento do registro do produto. Além disso, em caso de fiscalização ou investigação posterior, a ausência de registro de notificação agrava significativamente a situação da empresa.
Queixas técnicas que não causaram dano precisam ser notificadas?
Sim, desde que o desvio seja relevante. A RDC 67/2009 exige a notificação de queixas técnicas que, mesmo sem causar dano imediato, representem falha que poderia ter causado dano em outras circunstâncias ou que comprometam as especificações essenciais do produto. O prazo geral é de 30 dias. Pequenas reclamações estéticas ou de embalagem secundária, sem impacto funcional, geralmente não se enquadram nessa obrigatoriedade, mas devem ser registradas internamente.
Como funciona o e-NOTIVISA e qual é o impacto para fabricantes?
O e-NOTIVISA é uma plataforma experimental da ANVISA que permite que cidadãos registrem ocorrências com produtos de saúde e se comuniquem diretamente com as empresas fabricantes ou importadoras. Para fabricantes, isso significa que podem receber notificações de usuários finais diretamente pelo sistema, aumentando o volume e a diversidade de informações de campo disponíveis para análise. É importante monitorar essas comunicações e integrá-las ao processo de tecnovigilância da empresa.
Um produto importado está sujeito às mesmas obrigações de tecnovigilância que um produto nacional?
Sim. O importador detentor de registro no Brasil assume integralmente as obrigações de tecnovigilância para o produto que distribui no mercado nacional. Isso inclui notificar eventos adversos ocorridos no Brasil, implementar ações de campo quando necessário e manter o sistema de PMS ativo. Mesmo que o fabricante estrangeiro já tenha notificado o evento em seu país de origem, o importador brasileiro deve notificar à ANVISA de forma independente, dentro dos prazos brasileiros.
Próximos Passos para Estruturar seu Programa de Tecnovigilância
Para empresas que ainda não possuem um programa estruturado, recomendamos a seguinte sequência de implementação:
- Mapeie seu portfólio: identifique todos os produtos com registro ANVISA e classifique-os por classe de risco (I, II, III).
- Designe um responsável: nomeie formalmente um Responsável Técnico de Tecnovigilância com acesso ao NOTIVISA e conhecimento da RDC 67/2009.
- Elabore os POPs: documente os fluxos de recebimento, triagem, notificação e encerramento de ocorrências.
- Cadastre-se no NOTIVISA: faça o cadastro da empresa no sistema e familiarize-se com as funcionalidades antes de precisar usar em caráter de urgência.
- Implante o sistema de rastreabilidade: garanta que cada lote distribuído possa ser localizado em até 24 horas, com contatos de todos os compradores diretos.
- Realize uma simulação anual: execute um exercício de recall simulado para testar o tempo de resposta e identificar falhas no processo antes que um evento real ocorra.
A tecnovigilância não é apenas uma obrigação regulatória, é um sistema de inteligência que, quando bem implementado, fornece dados valiosos para a melhoria contínua dos produtos e para a demonstração proativa de segurança junto a compradores hospitalares, operadoras de planos de saúde e autoridades regulatórias. Empresas com programas maduros de pós-mercado são percebidas como parceiras mais confiáveis e têm vantagem competitiva em processos de licitação pública e credenciamento hospitalar.
Para aprofundar os aspectos de gestão de risco que fundamentam a tecnovigilância, consulte nosso artigo sobre a ISO 14971 e gestão de risco em dispositivos médicos. Para entender o contexto regulatório mais amplo, acesse o guia completo sobre dispositivos médicos na ANVISA em 2026.
Fontes e referências externas: NOTIVISA, Portal ANVISA Tecnovigilância ANVISA IMDRF, International Medical Device Regulators Forum FDA MDR Requirements EU MDR Guidance, European Commission.
Artigos relacionados
ANVISA e Dispositivos Médicos: Guia Completo de Regulamentação [Atualizado 2026]
Guia completo sobre regulamentação de dispositivos médicos pela ANVISA: classes de risco I-IV, fluxo de registro, documentação exigida, prazos, custos e as principais RDCs de 2022 a 2025. Conteúdo atualizado para profissionais de regulatory affairs, startups e fabricantes.
Certificação INMETRO para Equipamentos Médicos: Processo Passo a Passo
Guia completo sobre a certificação INMETRO de equipamentos eletromédicos no Brasil: Portaria 384/2020, normas IEC 60601, laboratórios acreditados, custos, prazos e o processo detalhado passo a passo para fabricantes e importadores.
Como Registrar um Dispositivo Médico na ANVISA: Roteiro Prático para Startups e Fabricantes
Guia completo do processo de registro de dispositivos médicos na ANVISA: classificação, CBPF, ensaios técnicos, submissão no sistema SOLICITA e acompanhamento. Prazos reais, custos atualizados e erros mais comuns a evitar.
IEC 60601: A Norma de Segurança de Equipamentos Eletromédicos Que Todo Engenheiro Precisa Conhecer
Entenda a estrutura completa da IEC 60601, o modelo sanduíche de normas gerais, colaterais e particulares, os conceitos de MOPP/MOOP, classificação de partes aplicadas e os requisitos de ensaio que definem a segurança de equipamentos eletromédicos no Brasil e no mundo.